|
|
Поліелектроліти та їх розчини.
Полімери, які містять в основному або бічних ланцюгах іоногенні групи, що здатні до дисоціації, носять назву поліелектролітів. Аналогічно низькомолекулярним електролітам поліелектроліти поділяються на полікислоти, які здатні в водному розчині відщепляти протони, тобто в склад їх макромолекул входять кислотні групи, наприклад, ─COOН, ─OSO3Н, поліоснови, які здатні в водному розчині приєднувати протони з утворенням полікатіонних макромолекул, які містять функціональні групи ─NH3+, =NH2+, інші, та поліамфоліти, макромолекули яких включають як кислотні, так і основні групи. Ці групи в процесі дисоціації компонентів розчину перетворюються відповідно в аніонні та катіонні групи.
Поліелектроліти, за виключенням білків, характеризуються високою щільністю розміщення іоногенних груп - на одну ланку ланцюга припадає по одній іоногенній групі. У білків , як представників поліамфолітів, одна карбоксильна група або аміногрупа зазвичай припадає лише на 6-8 залишків амінокислот.
Системи поліелектроліт - розчинник можуть бути двох типів:
- рідкими, у вигляді окремих макромолекул в розчині;
- твердоподібними, у вигляді драглю, коли поліелектроліт являє собою суцільну матрицю.
Всі незшиті поліелектроліти розчинні у полярних розчинниках, найкраще у воді, внаслідок сильної взаємодії іоногенних груп макромолекул з полярними рідинами.
Через наявність заряду макромолекули поліелектролітів відчувають у водних розчинах значні електростатичні взаємодії, які викликають значну деформацію їх ланцюгів, якщо макромолекули гнучкі. Цю деформацію визначає ступінь іонізації іоногенних груп, яка в свою чергу залежить від присутності в системі низькомолекулярних електролітів, рН середовища та ряду інших факторів. Наприклад, при зміні умов існування водного розчину (зміні рН) ланцюги поліакрилової кислоти можуть легко розтягуватися і скорочуватися у декілька разів.
Розчини поліелектролітів, як правило, відносять до істинних розчинів, проте за рядом властивостей (нерівномірний розподіл зарядів та маси розчиненої речовини, тощо) такі розчини ближче до колоїдних.
В водному розчині макромолекула поліелектроліту перетворюється в полііон. Особливість полііону полягає в тому, що його функціональні групи, які дисоціюють, пов’язані між собою хімічними зв’язками. Полііон в розчині оточений еквівалентною кількістю протилежно заряджених іонів (протиіонів). Протилежно заряджені іони малі і на декілька порядків менші за розміром ніж полііон.
Макромолекули поліелектролітів суттєво відрізняються від молекул низькомолекулярних електролітів і макромолекул неелектролітів. Від молекул низькомолекулярного електроліту макромолекула поліелектроліту відрізняється тим, що заряди полііону фіксовані і не здатні бути самостійними внаслідок зв’язку з ланцюгом.
 Макромолекула поліелектроліту в порівнянні з макромолекулою неелектроліту обмежена в можливості приймати різноманітні конформації внаслідок дії електростатичних сил. Особливо це проявляється в сильнозаряджених поліелектролітах, де ці електростатичні сили діють на відносно великих відстанях і значно перевищують дисперсійні та диполь-дипольні взаємодії. Потенційна енергія кулонівської взаємодії (Еп) двох заряджених ланок i та j , що знаходяться на відстані rij одна від одної, дорівнює:
Макромолекула поліелектроліту в порівнянні з макромолекулою неелектроліту обмежена в можливості приймати різноманітні конформації внаслідок дії електростатичних сил. Особливо це проявляється в сильнозаряджених поліелектролітах, де ці електростатичні сили діють на відносно великих відстанях і значно перевищують дисперсійні та диполь-дипольні взаємодії. Потенційна енергія кулонівської взаємодії (Еп) двох заряджених ланок i та j , що знаходяться на відстані rij одна від одної, дорівнює:
Еп = (e2/ D rij ) exp (- rij / rD ) ,
де e – заряд ланки, D – діелектрична проникність розчинника, rD – так званий дебаєвський радіус, що обумовлений екрануванням електростатичної взаємодії іонами, що знаходяться у розчині. Схема зарядженої макромолекули поліоснови наведена на рис.48.
Якщо полікислота чи поліоснова слабо дисоціює в водному розчині її відносять до слабких. Заряд макромолекул слабких полікислот та поліоснов визначається константами дисоціації іоногенних груп і суттєво залежить від рН розчину. Простою слабкою синтетичною полікислотою є поліакрилова кислота, яка слабо дисоціює в водному розчині:

До слабких полікислот відносяться також поліметакрилова, поліглутамінова, полігалактурова, альгінова і деякі інші кислоти.
Прикладом простої синтетичної поліоснови є полівініламін – слабкий поліелектроліт:

В сольову форму полівініламін переходить тільки в сильнокислому середовищі по схемі:
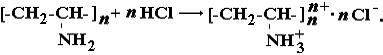
До слабких поліоснов відносять і інші полімери, які містять атоми азоту, що здатні приєднувати протон в водному розчині. Прикладами можуть бути полі -4- вінілпиридин, полі - п – аміностирол, поліетиленімін ( з вторинними атомами азоту в основному ланцюгу).
Сильні полікислоти, наприклад, полістиролсульфонова кислота, дисоціюють в водному середовищі практично повністю при любому значенні рН:

До цього класу кислот відносяться також поліетиленсульфокислота, гепарин, поліфосфорна кислота, іоногені групи якої розташовані в основному ланцюгу.
Сильною поліосновою є полівініл –п – толілтриметиламоній:

Іншими прикладами сильної поліоснови є полі -4- вініл-N-етилпірідіній бромід, полігесаметиленгуанідин солянокислий.
Сильні поліоснови з четвертинними атомами азоту в основному ланцюгу носять назву іоненів, наприклад:

Ці полімери стійкі в водних розчинах у вигляді вільних поліоснов.
Якщо в макромолекулах знаходяться четвертинні атоми фосфору чи третинні атоми сірки, то ці полімери теж проявляють властивості сильних поліоснов.
Поліелектроліти, що містять в макромолекулах кислотні та основні групи, носять назву поліамфолітів. До цих сполук відноситься ряд синтетичних кополімерів, а також білки. Прикладом синтетичного поліамфоліту може бути кополімер метакрилової кислоти з 4- вінілпиридином.
Білки дуже важливі з біологічної точки зору. Будову білка можна представити формулою:

де R1, R2, R3, … Ri – залишки L – амінокислот. Білки побудовані головним чином з залишків 20 амінокислот в різних з’єднаннях. Ці залишки амінокислот можуть бути неполярними (наприклад залишок лейцину -CH3-CH(CH3)-CH3),
так і полярними, в склад яких входять іоногенні групи, які володіють основними (наприклад, залишок лізіну –СН2–СН2–СН2–СН2-NH2), або кислотними (наприклад, залишок глютамінової кислоти –СН2–СН2–СООН) властивостями.
Дисоціацію іоногенних груп в білках можна проілюструвати на прикладі елементу поліпептидного ланцюга білкової молекули, який утворений двома амінокислотами – лізіном та аспарагіновою кислотою:
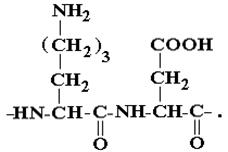
В водному розчині кислотні (- СООН) та основні ( - NH2) групи частково іонізовані; ступені їх дисоціації визначаються силою відповідних груп:
 .
.
Зміна рН розчину призводить до зміни ступеня дисоціації груп, причому надлишок іонів водню подавлює дисоціацію карбоксильної групи і сприяє іонізації аміногрупи, а надлишок гідроксид-іонів подавлює іонізацію аміногрупи і сприяє дисоціації карбоксильної групи:

 Зміною рН середовища можна досягти такого стану, коли кількість позитивних та від’ємних зарядів на кожній макромолекулі буде однаковою, т.б. сумарний заряд макромолекули дорівнював нулю. Такий стан носить назву ізоелектричного, а значення рН, при якому кількість іонізованих кислотних та основних груп стає однаковою, ─ ізоелектричною точкою
Зміною рН середовища можна досягти такого стану, коли кількість позитивних та від’ємних зарядів на кожній макромолекулі буде однаковою, т.б. сумарний заряд макромолекули дорівнював нулю. Такий стан носить назву ізоелектричного, а значення рН, при якому кількість іонізованих кислотних та основних груп стає однаковою, ─ ізоелектричною точкою
(ІЕТ). В розчинах з рН < pHІЕТ макромолекули мають надлишковий позитивний, а при рН > pHІЕТ ─ від’ємний заряд.
Якщо водний розчин поліамфоліту містить окрім кислоти або лугу ще й інші електроліти, то специфічне зв’язування іонів цих електролітів може суттєво змінити поглинання поліамфлітом ( в тому числі і білком ) іонів гідроксиду або водню. Тому, поліамфоліти характеризують ще й ізоіонною точкою ─ значенням рН, при якому поліамфоліт поглинає із розчину рівну кількість іонів Н+ та ОН-, тобто сумарний заряд іонів Н+ та ОН, що поглинуті, дорівнює нулю.
У відсутності сторонніх електролітів (окрім кислот та лугів ) ізоелектрична та ізоіонна точки співпадають. При введенні в розчин іонів, що специфічно зв’язуються, цього не відбувається. Якщо поліамфоліт зв’язує переважно аніони, то частина іонів ОН- витісняється з поліамфоліту в розчин, ізоелектричний стан досягається в більш лужному середовищі, а положення ізоелектричної точки зміщується в бік більш високих значень рН; при специфічному зв’язуванні катіонів в розчин виділяються іони Н+ і положення ізоелектричної точки зміщується в бік менших значень рН.
Положення ізоелектричної точки поліамфоліту визначається силою кислотних та основних груп та їх відносною кількістю. Для білків найчастіше кислотні властивості превалюють над основними і їх ізоелектрична точка розташована в області малих значень рН. Це ілюструє табл.1.
 В поліелектролітних драглях полімерна сітка несе фіксовані заряди одного знаку, які врівноважені рухомими протиіонами. В якості одиничного полііону в цих драглях розглядається один фіксований іон разом з частиною сітки, що припадає на цей іон, хоча можна вважати і всю просторову сітку одним великим полііоном.
В поліелектролітних драглях полімерна сітка несе фіксовані заряди одного знаку, які врівноважені рухомими протиіонами. В якості одиничного полііону в цих драглях розглядається один фіксований іон разом з частиною сітки, що припадає на цей іон, хоча можна вважати і всю просторову сітку одним великим полііоном.
Є певні загальні закономірності поведінки обох типів поліелектролітних систем, які можуть бути пояснені за допомогою моделей системи поліелектроліт – зовнішній розчин, рис.49.
В перший моделі полііони (R─) можуть рухатися в об’ємі 1, але не можуть проникати в об’єм 2, де знаходиться розчин низькомолекулярного електроліту К+А─ (К+ - катіон, А─ - аніон), оскільки об’єми 1 та 2 відокремлені напівпроникною мембраною. Остання проникна для молекул розчинника, катіонів та аніонів , але непроникна для полііонів. В даному випадку полііон несе від’ємний заряд, а протиіон К+ є загальним для поліелектроліту та низькомолекулярного електроліту.
Друга модель уособлює систему поліелектролітний драгль - зовнішній розчин і не включає мембрани. В об’ємі 1 також рівномірно розподіляються полііони і рухомі протиіони. Відміна цієї моделі від попередньої полягає в тому, що полііони в ній нерухомі внаслідок фіксації їх в сітці.
Загальним для обох моделей є локалізація полііонів в об’ємі 1 і вільне переміщення по об’ємам 1 та 2 іонів К+ та А─, які розподіляються між цими об’ємами.
Для визначення закону рівноважного розподілу іонів К+ та А─ між обома об’ємами будемо вважати, що ці об’єми є однаковими. Умова рівноваги в таких системах (при постійних зовнішнього тиску та температурі) повинна бути виражена рівністю не хімічних, а електрохімічних потенціалів двох фаз, що контактують, (  ), тобто:
), тобто:
 ,
,
де mi - хімічний потенціал, а  - електрична компонента енергії, яка дорівнює добутку заряду q на потенціал φ, що має розмірність енергії. Для мольної енергії Гібса електрична компонента дорівнює , де zi – заряд іону (включаючи знак), F – число Фарадея. Тоді:
- електрична компонента енергії, яка дорівнює добутку заряду q на потенціал φ, що має розмірність енергії. Для мольної енергії Гібса електрична компонента дорівнює , де zi – заряд іону (включаючи знак), F – число Фарадея. Тоді:
 .
.
В цьому випадку рискою позначений електрохімічний потенціал системи в об’ємі 1.
Потенціал ψ відраховується від нульового рівня, який відповідає глибині розчину на відстані х (х=∞), ψ= (ψ)х – (φ)х=∞ .
Врахуємо, що:
 ,
,
де активність а дорівнює концентрації с (аi=ci) при с→ 0, а  . Тоді для бінарного електроліту (z+=─z_ =z) , знаходимо:
. Тоді для бінарного електроліту (z+=─z_ =z) , знаходимо:

 .
.
Складаючи ці обидва рівняння одержимо основний вираз для іонної рівноваги:
 .
.
Це рівняння носить назву мембранної або донанівської рівноваги.
Якщо коефіцієнти активності рухомих іонів в об’ємах 1 та 2 рівні, то можна записати, що
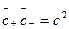 . (35)
. (35)
Оскільки склад рідин змінюється за рахунок дифузії малих іонів, причому електронейтральність в кожному об’ємі зберігається, то рівняння (35) можна представити у вигляді:
 . (36)
. (36)
де  початкова концентрація катіонів в об’ємі 1,
початкова концентрація катіонів в об’ємі 1,  та
та  концентрації катіонів та аніонів, що перейшли з об’єму 2 в об’єм 1, с0 – вихідна концентрація кожного з іонів низькомолекулярного електроліту в об’ємі 2, а сх – концентрація кожного з іонів, що перейшли з об’єму 2 в об’єм 1.
концентрації катіонів та аніонів, що перейшли з об’єму 2 в об’єм 1, с0 – вихідна концентрація кожного з іонів низькомолекулярного електроліту в об’ємі 2, а сх – концентрація кожного з іонів, що перейшли з об’єму 2 в об’єм 1.
Вирішення рівняння (36) дає:
 . (37)
. (37)
В розбавленому розчині поліелектроліту або малому вмісту полімерного драгля справедлива нерівність << c0 , тобто в системі дуже мала кількість полімерної речовини, що не здатна до дифузії через границю між об’ємами 1 та 2. В цьому випадку можна знехтувати доданком в знаменнику: Тоді:
 .
.
Це означає, що в стані рівноваги низькомолекулярний електроліт розподілений рівномірно по обидві сторони границі між об’ємами 1 та 2.
Якщо ≈с0 , то :
 ,
,
а це вказує, що концентрації низькомолекулярного електроліту в об’ємах 1 та 2 в стані рівноваги нерівні і ця нерівність збільшується по мірі підвищення концентрації речовини (поліелектроліту) , що не здатний до дифузії через границю об’ємів, – знаменник в рівнянні (37) збільшується, тобто проходження низькомолекулярного електроліту через границю об’ємів зменшується.
Присутність низькомолекулярного електроліту в об’ємі 2 змінює осмотичний тиск, який створює присутність в розчині поліелектроліту. Для аналізу такої зміни запишемо рівняння Вант-Гофа для осмотичного тиску розчину поліелектроліту (об’єм 1 ), що контактує з розчином низькомолекулярного електроліту (об’єм 2), у формі:
 , (38)
, (38)
де  , оскільки в рівнянні Вант-Гофа треба врахувати концентрацію полііонів
, оскільки в рівнянні Вант-Гофа треба врахувати концентрацію полііонів  , а кожний полііон дисоціює з утворенням z протиіонів. Після розкриття скобок і підстановки значення сх з рівняння (37), одержимо:
, а кожний полііон дисоціює з утворенням z протиіонів. Після розкриття скобок і підстановки значення сх з рівняння (37), одержимо:
 , (39)
, (39)
Аналіз рівняння (39) показує, що при >>c0
 ,
,
тобто осмотичний тиск визначається електролітом, що продисоціював.
При <<c0

і осмотичний тиск визначається поліелектролітом, дисоціація макромолекул якого подавлена.
Мембранна рівновага і рівновага на межі полімерний драгль –зовнішній розчин відіграють дуже важливу роль в життєдіяльності живих організмів, зокрема в процесах обміну речовин.
Рівноважні властивості розчинів поліелектролітів суттєво відрізняються від ідеальних орозчинів. Так, осмотичний тиск π безсольових розчинів поліелектролітів значно нижчий, ніж очікуваний для ідеального розчину (πід). Мірою цієї різниці є величина осмотичного коефіцієнту
Фр = π/πід ,
який для поліелектролітів не залежить від їх молекулярної маси та концентрації в розчині, але помітно зменшується при зростанні лінійної щільності заряду. Для типових поліелектролітів вінілового ряду Фр ~ 0,1.
Основною причиною відхилення поведінки розчинів поліелектролітів від ідеального розчину є вплив сильного електричного поля полііону на характер розподілу протиіонів. При високій щільності заряду частина протиіонів зв’язується з полііоном, щоб знизити цю щільність заряду до певної критичної величини. Остання співпадає з так званою зворотною величиною бьєррумовської довжини :
e2 / D kT,
де е - заряд протону, D - діелектрична проникність розчинника, k - стала Больцмана, Т - абсолютна температура.
Показником , який характеризує зв’язування протиіонів, є безрозмірний критичний параметр x:
x = (e2 / DkT ) / b ,
де b - проекція відстані між сусідніми зарядженими групами полііону на вісь повністю витягнутого ланцюга. При х >1 певна доля протиіонів зв’язується з полііоном. Вона дорівнює 1─ x ─1. Внаслідок цього x досягає свого критичного значення, що дорівнює одиниці. В протилежному випадку система буде термодинамічно нестійкою.
На полііоні з низькою щільністю заряду (х<1) зв’язування протиіонів не відбувається.
Властивості системи поліелектроліт-розчинник суттєво залежать від рН розчину, а для поліелектролітних драглів – рН зовнішнього розчину.
Так, набухання поліелектролітів у воді сильно залежить від рН середовища, рис.8.
Для полікислот та поліоснов (рис.8, криві 1 та 2) відповідно з зростанням або зменшенням рН набухання збільшується, що пов’язано з іонізацією відповідних іоногенних груп. В області великих ( для полікислоти) і малих ( для поліоснови) рН спостерігається деяке зменшення набухання, що пов’язане з подавленням іонізації однойменно заряджених іоногенних груп.
Для поліамфолітів і ,зокрема, білків в ізоелектричній точці макромолекула поліамфоліту згортається в щільний клубок і тому набухання мінімальне (рис.8,крива 3). Поява надлишкового заряду якого-небудь знаку при збільшенні або зменшенні рН сприяє розгортанню макромолекул за рахунок відштовхування однойменних зарядів і набухання збільшується. В області великих та малих значень рН набухання поліамфоліту дещо зменшується, що пов’язано, як і у випадку індивідуальних полікислот та поліоснов, з збільшенням іонної сили розчину, в якому відбувається набухання.
 Типовий вигляд кривих, що наведені на рис.8, тобто залежностей ступеня набухання як функція рН середовища для різних поліелектролітів, мають і залежності від рН характеристичної в’язкості розчинів поліелектролітів [η], проходження через них світла (Iпр), осмотичного тиску розчинів (π), електрофоретичної рухомості макромолекул (υ), рис.50. Зокрема при електрофорезі швидкість руху макромолекул пропорційна напрузі електричного поля, що прикладається (Е):
Типовий вигляд кривих, що наведені на рис.8, тобто залежностей ступеня набухання як функція рН середовища для різних поліелектролітів, мають і залежності від рН характеристичної в’язкості розчинів поліелектролітів [η], проходження через них світла (Iпр), осмотичного тиску розчинів (π), електрофоретичної рухомості макромолекул (υ), рис.50. Зокрема при електрофорезі швидкість руху макромолекул пропорційна напрузі електричного поля, що прикладається (Е):
υ = КЕ ,
де К- коефіцієнт електрофоретичної рухомості, і при умові, що υ=0, яка досягається зміною рН середовища, коефіцієнт К теж дорівнює нулю, тобто досягнутий стан ізоелектричної точки.
 Різна здатність макромолекул поліелектролітів і ,зокрема, біологічних поліелектролітів (білків) з переважним зарядом якого-небудь знаку, яка обумовлена різною величиною заряду, пересуватися в електричному полі дає змогу провести розподіл індивідуальних білків з їх суміші . Експериментальні данні одержують у вигляді електрофореграми – кривої, яка відображує залежність концентрації компонентів суміші п від відстані h, рис.51.
Різна здатність макромолекул поліелектролітів і ,зокрема, біологічних поліелектролітів (білків) з переважним зарядом якого-небудь знаку, яка обумовлена різною величиною заряду, пересуватися в електричному полі дає змогу провести розподіл індивідуальних білків з їх суміші . Експериментальні данні одержують у вигляді електрофореграми – кривої, яка відображує залежність концентрації компонентів суміші п від відстані h, рис.51.
Кожен пік відповідає певному компоненту суміші. Площа під піком характеризує вміст компоненту у розчині.
Здатність білкових макромолекул змінювати конформацію при зміні рН середовища лежить в основі ферментативного каталізу, який відбувається на молекулярному рівні в живих організмах. Схема утворення центру активності для  ферментативного каталізу наведена на рис.52.
ферментативного каталізу наведена на рис.52.
Центр активності ферменту здатний утворювати з речовиною, яку потрібно перетворити (субстрат ), ферментативно-субстратний комплекс внаслідок відповідності структури (рельєфу) обох речовин. Механізм каталізу на молекулярному рівні запропонований Полінгом і носить назву механізму ключа та замкової
шпари.
Прикладом ферментативного каталізу, що відбувається на молекулярному рівні, є дія трипсину на желатин.
Наявність в макромолекулах поліамфолітів – білків функціональних груп, що здатні при певному значені рН до дисоціації з утворенням дисоційованих груп типу -СОО-, -NH3+, -OH-, сприяє розчиненню поліамфоліту у воді. Інші радікали, зокрема –СН3, бензольні кільця, надають макромолекулам гідрофобності. Зокрема, гідрофобні амінокислоти ,що входять до складу макромолекул білків, наприклад,
 (гліцин) ,
(гліцин) ,  (аланін)
(аланін)
нерозчинні у воді, не вбудовуються в а- спіраль Полінга – Корі іпослаблюють стійкість спіралі у водному розчині.
Гідрофільні амінокислоти, які теж часто входять до складу макромолекул білків, наприклад,
 (серін)
(серін)  (тірозін),
(тірозін),
 завдяки групі –ОН розчинні у воді.
завдяки групі –ОН розчинні у воді.
Ефективний об’єм (Vеф), який займає макромолекула білка у воді, буде залежати від кількісного співвідношення гідрофобних та гідрофільних радікалів (m), що містить макромолекула білка. Це ілюструє діаграма Фішера, яка наведена на рис.53.
В водному розчині макромолекула білка намагається згорнутися, але просте згортання часто неможливе. В формі α –спіралі Полінга та Корі макромолекула білків стабілізується внутрішньомолекулярними водневими зв’язками та диполь-дипольними взаємодіями. β- структура білків стабілізується міжмолекулярними водневими зв’язками, що веде до паралельного розташування витягнутих до максимальної межі макромолекул і не дає останнім можливості згорнутися, прийняти форму клубку. Останнє, наприклад, використовує шовкопряд витягуючи (а не видавлюючи) вміст своєї залози, що призводить до того, що слабо згорнуті макромолекули фіброїну орієнтуються і утворюється β- структура білку. Фіброїн, а також кератин (вовна) займають ліву частину діаграми Фішера.
Казеїн, що виділяється з молока, займає праве положення діаграми Фішера. При контакті з водою його макромолекули, що попередньо силовим полем були розпрямлені і стабілізовані в β- структуру, дістають великий надлишок енергії, що веде до подолання сил міжмолекулярної взаємодії. Ці міжмолекулярні зв’язки порушуються і макромолекули одержують можливість прийняти конформації, що відповідають правій частині діаграмі Фішера. Тепер, якщо з такого білку була створена тканина, то при контакті з вологою буде спостерігатися її значний збіг та розповзання.
Зміною рН оточуючого середовища можна перетворити плівку поліелектроліту в хімічну машину (або хімічний м’яз) згідно схеми, що наведена на рис.54. Змінимо рН середовища від деякого значення кислого або лужного середовища до значення , що відповідає ізоелектричній точці, в якій макромолекула  вважається в цілому електронейтральною, хоча вона і має іонізовані групи. Тоді внаслідок гнучкості і намагання перейти в стан з мінімумом вільної енергії макромолекула згорнеться в клубок. Щільність клубка внаслідок дії сил притягання між різнойменно зарядженими групами буде більшою від тієї щільності, котра відповідає найбільш статистично вірогідній формі макромолекули або максимальній її ентропії. Таким чином, зміною рН середовища досягається зміна відстані між кінцями ланцюгів і виконується механічна робота по підняттю ваги (Р). Подальша зміна рН середовища призведе до опускання цієї ваги.
вважається в цілому електронейтральною, хоча вона і має іонізовані групи. Тоді внаслідок гнучкості і намагання перейти в стан з мінімумом вільної енергії макромолекула згорнеться в клубок. Щільність клубка внаслідок дії сил притягання між різнойменно зарядженими групами буде більшою від тієї щільності, котра відповідає найбільш статистично вірогідній формі макромолекули або максимальній її ентропії. Таким чином, зміною рН середовища досягається зміна відстані між кінцями ланцюгів і виконується механічна робота по підняттю ваги (Р). Подальша зміна рН середовища призведе до опускання цієї ваги.
Схема хімічного м’язу запропонована Качальським. Прикладом поліелектроліту, здатного виконати роль штучного м’язу, є кополімер акрилової кислоти з вініловим спиртом.
Якщо вага (Р) занадто велика, то плівка поліелектроліту викидає іони в оточуюче середовище змінюючи його рН. Тобто, спостерігається взаємозв’язок причини та наслідку. При малій вазі змінюється відстань між кінцями ланцюгів, при великій вазі – змінюється рН середовища.
Електрохімічні властивості розчинів поліелектролітів.
Електрохімічні властивості поліелектролітів відрізняються від властивостей розчинів низькомолекулярних електролітів. Полімерні кислоти та основи значно слабкіші від своїх низькомолекулярних аналогів.
При дисоціації слабкого поліелектроліту по аналогії з дисоціацією молекул слабкого низькомолекулярного електроліту в розчині полімеру встановлюється рівновага між кількістю іоногенних груп макромолекул, що зазнали дисоціації, і кількістю іоногенних, що не продисоціювали. Для со моль-ланок/л ланцюга (кожна мономерна ланка має одну іоногенну групу) такого поліелектроліту, наприклад, слабкої полікислоти, при дисоціації іоногенних груп з ступенем іонізації а ( а=[H+] /c0, оскільки концентрація протонів[H+] дорівнює концентрації аніонів ланцюга [A-], які утворилися при дисоціації іоногенних груп ), 1-а молів іоногенних груп залишається не продисоційованними. Тому концентрація іоногенних груп, що не продисоціювали [HA ], дорівнює:
[HA ] = c0(1-a) .
Введемо величини [H+] та [HA ] в вираз для константи дисоціації слабкої полікислоти КD , яка може бути записана рівнянням:

і одержимо:
 . (40)
. (40)
Рівняння (40), яке справедливо для любих слабких бінарних низькомолекулярних електролітів так і поліелектролітів, носить назву закону розведення Освальда . Воно може бути записане також у вигляді:
 або
або  . (41)
. (41)
Для слабких низькомолекулярних електролітів константа дисоціації є характеристичною величиною і не залежить від ступеня іонізації електроліту, а також концентрації. Ця характеристична величина пов’язана з зміною стандартної вільної енергії Гібса іонізації молекул (∆G0) рівнянням:
 , (42)
, (42)
де В=0,434, а КD,0 – характеристична константа дисоціації слабкого низькомолекулярного електроліту.
Для поліелектроліту КD не є характеристичною величиною і залежить від ступеню іонізації, рис.55. Причиною цього є те, що здатність полікислоти відщеплювати протон, а поліоснов приєднувати його послаблюється при збільшенні α (тобто ступеню іонізації) через прогресуюче зростання кулонівської взаємодії між полііоном та протиіонами. Наприклад, для полікислоти протон, що утворюється внаслідок дисоціації іоногенної групи, зі зростанням α відчуває все більше притягання з боку полііону і робота відриву протону зростає.
 Цей ефект суттєво зменшується при введенні в розчин простих солей (індиферентних електролітів), що екранують заряди полііонів, і при збільшенні концентрації простих солей електрохімічна поведінка розчинів слабких поліелектролітів наближається до поведінки низькомолекулярних аналогів.
Цей ефект суттєво зменшується при введенні в розчин простих солей (індиферентних електролітів), що екранують заряди полііонів, і при збільшенні концентрації простих солей електрохімічна поведінка розчинів слабких поліелектролітів наближається до поведінки низькомолекулярних аналогів.
Особливістю застосування рівняння (42) для поліелектроліту є те, що в нього вводиться додаткова робота (додаткова електростатична вільна енергія ∆Gел.), яка потрібна для відокремлення еквівалентної кількості протонів від полііону в нескінченність. Тоді:
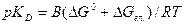
і рівняння (41) може бути записано у вигляді:
 .
.
При зміні заряду (z) макромолекули від (z) до (z+1), тобто коли в додаток до існуючих іоногенних груп, що продисоціювали, додатково продисоціювала ще одна іоногенна група
∆Gел=∂∆Gел.(z) / ∂z
і при ступені дисоціації іоногенних груп а
 ,
,
де ступінь дисоціації а дорівнює a=z/N, а N – загальне число іоногенних груп в макромолекулі.
Для опису електрохімічної поведінки поліелектролітів найчастіше користуються значенням "характеристичної" константи дисоціації КD,0, тобто дисоціації одиничної іоногенної групи у відсутності інших заряджених груп в ланцюзі. Її отримують екстраполяцією pКD до нульового значення α. Значення КD,0 для поліелектроліту та відповідного низькомолекулярного аналога, як правило, близькі, хоч вони не співпадають, тому що іоногенна група в полімерному ланцюзі знаходиться в іншому мікрооточені ніж іони в розчині.
Величина ∆Gел залежить від іонної сили розчину J, яка дорівнює сумі добутків концентрації на квадрат валентності для усіх іонів, що знаходяться в розчині
 .
.
 Збільшення іонної сили розчину призводить до послаблення електростатичних взаємодій в полімерному ланцюгу внаслідок часткового екранування зарядів і, відповідно, до зменшення ефективної константи дисоціації слабкої полікислоти. Це ілюструють криві потенціометричного титрування поліакрилової кислоти в присутності різної кількості хлористого калію, що наведені на рис.56.
Збільшення іонної сили розчину призводить до послаблення електростатичних взаємодій в полімерному ланцюгу внаслідок часткового екранування зарядів і, відповідно, до зменшення ефективної константи дисоціації слабкої полікислоти. Це ілюструють криві потенціометричного титрування поліакрилової кислоти в присутності різної кількості хлористого калію, що наведені на рис.56.
Внаслідок збільшення іонної сили розчину крива титрування поліелектроліту наближається до кривої титрування низькомолекулярного аналогу. Однак, навіть при значній іонній силі розчину криві для поліелектроліту та його низькомолекулярного аналогу не співпадають, оскільки електростатична взаємодія між сусідніми іоногенними групами ланцюга не зникає.
Відомо, що криві титрування не залежать від довжини ланцюга полііону. Це вказує на те, що відстань між іоногенними групами, що продисоціювали, і взаємодія яких суттєво впливає на вільну електростатичну енергію, мало в порівнянні з розмірами полііону. Але крива титрування залежить від конфігурації ланцюга макромолекул та конформації останньої в розчині. Наприклад, для тієї ж поліакрилової кислоти перехід від ізотактичної до синдіотактичної супроводжується менш вираженою залежністю рК від а, тому що для останньої менша густина заряду вздовж ланцюга макромолекули.
Те, що було зазначено для слабкої полікислоти, характерно і для слабкої поліоснови з тією різницею, що по мірі дисоціації останньої збільшуються її кислотні властивості.
Дисоціація функціональних груп макромолекули поліелектроліту викликає електростатичне відштовхування між однойменно зарядженими ланками, що спричиняє розгортання ланцюга, який стає макроіоном. Зміна розміру полііону веде до зміни в розподілі фіксованих зарядів. Потенціал електростатичного поля для полііону, що має певну конформацію і , відповідно, певне розташування фіксованих зарядів, найчастіше розраховують за допомогою сферичної моделі, схема якої наведена на рис.57.

Найменш реалістичною є модель Куна, згідно якої вважається, що в дуже розведеному розчині протиіони виходять з поля полііону і не впливають на взаємне електростатичне відштовхування фіксованих зарядів. Однак, значна доля протиіонів завжди залишається в області полііону і екранує заряди іонів макроланцюга, що призводить до зменшення взаємного відштовхування останніх.
В моделі Качальского та Ліфсона вважається, що кожний фіксований заряд створює навкруги себе іонну атмосферу подібної до тої, яка утворюється в розчині низькомолекулярного електроліту. Опис цієї моделі базується на теорії розведених розчинів сильних низькомолекулярних електролітів. Розрахунок електричного потенціалу ψ іону, що знаходиться в оточенні інших іонів в розбавленому розчині сильного низькомолекулярного електроліту, зроблений Дебаем та Гюккелем. В їх теорії складна взаємодія багатьох іонів замінена більш простою взаємодією їх іонних атмосфер. До того дискретні заряди іонів всередині іонних атмосфер замінюються безперервним полем, густина заряду в кожному елементі об’єму якого пропорційна надлишковій концентрації іонів даного знаку в цьому елементі.
Іонна атмосфера, що екранує електростатичну взаємодію, вважається достатньо слабкою у порівнянні з інтенсивністю теплового руху іонів і приймається динамічною, що постійно змінюється, отже неможливо виділити в розчині деякий фіксований зв’язок між протилежно зарядженими іонами. Оскільки закон розподілу зарядів в іонній атмосфері відомий ( густина заряду в іонній атмосфері зменшується з відстанню від центрального іону по такому ж ступінному закону, як зменшується густина земної атмосфери з висотою), то може бути розрахований потенціал ψ в кожній її точці, включаючи точку, яку займає центральний іон, що розглядається. Для цього потенціалу розрахунок дає:
 , (43)
, (43)
а,  ,
,
де D – діелектрична постійна середовища, J – іонна сила, е – елементарний заряд, zi – валентність іону, N0 – кількість іонів в молі.
Вираз (43) відрізняється від закону Кулона для двох точкових зарядів лише тим, що відстань до другого заряду замінюється характеристичною довжиною 1/χ , яка може бути мірилом товщини іонної атмосфери.
ψ в даному випадку це не потенціал точкового заряду іону відносно другого точкового заряду, а потенціал іонної атмосфери в її центрі, де знаходиться даний іон.
Використовуючи вираз для електростатичної складової вільної енергії (∆Gел):
∆Gел=N0eψ ,
Качальский і Ліфсон вивели рівняння, які описують розміри полііону і термодинамічні властивості розчину.
Для поліелектролітів ситуація з іонними атмосферами принципово міняється, тому що необхідно враховувати взаємодію протиіону із зарядженим полімерним ланцюгом. Така взаємодія призводить до зв’язування частини протиіонів полімерним ланцюгом. Ефективний заряд сильних поліелектролітів створюють 10 - 20% мономерних ланок, тоді як заряди на решті 80-90% ланок компенсовані протиіонами. До того конформація макроіонів чутлива до присутності в розчині мультивалентних протиіонів. Введення таких протиіонів спричиняє згортання індивідуальних клубків або сприяє міжмолекулярній асоціації поліелектролітних ланцюгів.
В водних розчинах поліелектролітів має місце і конкуренція кулонівської та неелектростатичних взаємодій. Останні відносяться до гідрофобних взаємодій, які виникають внаслідок притягання неполярних фрагментів ланцюгів. Гідрофобні взаємодії особливо сильно проявляються у слабко заряджених поліелектролітів або електролітів, що містять об’ємні гідрофобні фрагменти. Наслідком є формування в розчині наноструктурних мікронеоднорідностей або мікродоменів.
Більш реалістичною є модель сфери, яка є проникною для розчинника, а також для малих іонів в область, яку займає полііон. Обмеженням в цій моделі є те ,що приймається рівномірний розподіл заряду в об’ємі, який займає полііон, і відсутність взаємодії між полііонами в розчині. Теоретичний опис такої моделі базується на вирішенні рівнянні Пуасона для безперервного електричного поля, яке зв’язує потенціал ψ в деякій точці х,у,z з густиною заряду ρ в цій точці:
 ,
,
де D – діелектрична постійна середовища і ∆ - оператор Лапласа.
 .
.
ψ в цій моделі визначається розташуванням фіксованих зарядів, розподіленням протиіонів, а також катіонів і аніонів додатково введеного електроліту.
У випадку коли фіксовані заряди є позитивно однозарядними і які рівномірно розподілені всередині сфери з концентрацією с і що полііон знаходиться в оточенні взятого в великому надлишку простого електроліту , наприклад, в співвідношенні 1:1, то:
 ,
,
де ε- заряд протону, сs – валова, тобто яка визначається аналітично, концентрація коіонів та проти іонів, χ – параметр Дебая, величина 1/χ, як зазначено вище, є характеристичною довжиною, або радіусом іонної атмосфери. Аналітичні рішення цього рівняння є різними.
Недосконалість сферичних моделей обумовлена заміною дискретних фіксованих зарядів поліону безперервним розподіленням заряду на поверхні або в об’ємі сфери. Тобто, приймається, що потенціал всередині сфери, що проникна, приблизно постійний. Однак, цей потенціал сильно змінюється від точки до точки, тобто його значення може давати значні позитивні та негативні відхилення від постійної величини яку задає теорія. А це призводить до більш ефективного екранування фіксованих зарядів реального полііону протиіонами і завищенню теоретичної оцінки розгортання цього полііону.
Реологія розчинів поліелектролітів.
Поліелектролітний ефект.
При дослідженні рівноважних властивостей розчинів поліелектролітів розрізняють безсольові розчини та розчини, що містять солі, які додатково введені. В першого типу розчинах вклад власне полііонів у рівноважні (коллігативні) властивості розчину поліелектроліту значно менший ніж вклад великої кількості протиіонів. Тому безсольові розчини не використовують для визначення молекулярної маси поліелектролітів.
Введенням в розчини поліелектролітів додаткових кількостей солей вдається з використанням адитивності для осмотичного тиску π в такій складній системі (π = πпе + πс, де πпе - виміряний осмотичний тиск безсольового розчину поліелектроліту, πс - осмотичний тиск розчину солі, що не містить поліелектроліту) виявити експериментально вклад полііонів у властивості розчину.
В розведених розчинах поліелектролітів спостерігається реологічний
ефект, який носить назву поліелектролітного . Він проявляється в зростанні
приведеної в’язкості розчину по мірі його розведення, рис.58. На цьому рисунку наведена концентраційна залежність приведеної в’язкості водного розчину полігесаметиленгуанідину солянокислого (ПГМГ) у відсутності індиферентного електроліту (крива 1) та в присутності – хлористого натрію (криві 2 та 3).
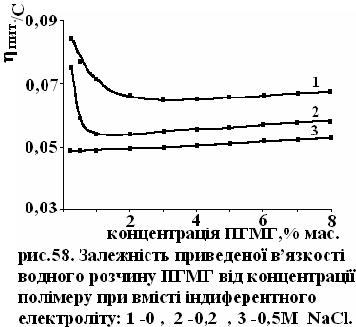 Причиною появи поліелектролітного ефекту є те, що при зменшенні концентрації поліелектроліту знижується іонна сила розчину, яка обумовлена незв’язаними з ланцюгом протиіонами. При розведенні розчину звільняється частина протиіонів, які були до цього зв’язані з макроіоном, ефективний заряд ланцюга підвищується і сили електростатичного відштовхування розгортають макромолекулярний клубок. Наслідком цього є розбухання макроклубків та збільшення гідродинамічних розмірів полііонів, що і призводить до зростання приведеної в’язкості розчинів поліелектроліту.
Причиною появи поліелектролітного ефекту є те, що при зменшенні концентрації поліелектроліту знижується іонна сила розчину, яка обумовлена незв’язаними з ланцюгом протиіонами. При розведенні розчину звільняється частина протиіонів, які були до цього зв’язані з макроіоном, ефективний заряд ланцюга підвищується і сили електростатичного відштовхування розгортають макромолекулярний клубок. Наслідком цього є розбухання макроклубків та збільшення гідродинамічних розмірів полііонів, що і призводить до зростання приведеної в’язкості розчинів поліелектроліту.
Поліелектролітний ефект описується рівнянням Фуоса:
 ,
,
де А – константа, яка залежить від молекулярної маси полімеру, а В – константа, яка пов’язана з структурою і визначає до якої міри розведення впливає на здатність макромолекул до розгортання.
Поліелектролітний ефект не виникає в розчинниках в яких неможлива дисоціація – органічних розчинниках з малою діелектричною проникністю. В водному розчині цей ефект подавлюється введенням в цей розчин певної кількості індиферентного електроліту, зокрема для розчину ПГМГ це введення 0,5М хлористого натрію, рис. 58.
У відсутності стороннього (індиферентного) електроліту конформація макроіона в дуже розведеному розчині наближається до стрижнеподібної. В таких умовах в рівнянні Марка- Куна-Хаувінка:
[η]=k М α ,
де [η] – характеристична в’язкість, k, – константа , показник α наближається до значення 2,0.
Іонна сила розчину регулюється додатковим введенням індиферентного електроліту. З збільшенням іонної сили показник а рівняння Марка- Куна-Хаувінка зменшує своє значення до 0,5. Це значення характерне для ідеальних макромолекул . І в розчині з дуже великою іонною силою конформація полііону наближається до конформації незбудженного статистичного клубка.
Для визначення характеристичної в’язкості поліелектроліту в умовах виключення поліелектролітного ефекту застосовують рівняння Ліберті-Стівела:
 ,
,
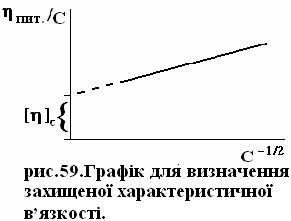 де
де  - носить назву захищеної характеристичної в’язкості. Останню визначають побудовою графічної залежності
- носить назву захищеної характеристичної в’язкості. Останню визначають побудовою графічної залежності  , рис.59.
, рис.59.
Іншій спосіб виключення поліелектролітного ефекту для визначення характеристичної в’язкості розчину полягає в розведенні розчину поліелектроліту розчинами низькомолекулярних електролітів такої концентрації, щоб іонна сила залишалася рівною іонній силі вихідного розчину поліелектроліту (ізоіонне розведення). Концентрацію низькомолекулярного електроліту встановлюють з досліду. При розведенні безсольових розчинів поліелектролітів найчастіше використовують розчин (1:1) валентного електроліту, нормальність якого дорівнює половині нормальності розчину поліелектроліту, що відповідає незмінній повній концентрації рухомих іонів.
З збільшенням іонної сили розчину значення характеристичної в’язкості зменшується, що пов’язане з зменшенням розміру полііону внаслідок блокування його фіксованих зарядів.
Реологічні властивості поліелектролітів в області напіврозбавлених і концентрованих розчинів досліджені в меншій мірі ніж розведені розчини. Так, область напіврозбавленого розчину без зачіплювань макромолекул була досліджена в розчинах полідиметилдиалліламоній хлориду і не виявлено аномалій в’язкості в концентраційному інтервалі до 35 мас.% при різних швидкостях зсуву.
Для опису концентрованих розчинів поліелектролітів і для концентрованих розчинів низькомолекулярних електролітів рівняння теорії Дебая і Гюккеля не придатні. Це пов’язано з дією додаткових факторів, якими можна було знехтувати в розведених розчинах. Це зміна діелектричної постійної розчинника поруч з полііонами та іонами, гідрофобні взаємодії, які істотно проявляються не тільки в концентрованих розчинах поліелектролітів, але і в концентрованих розчинах неелектролітів, можливе з’єднання іонів в пари і в більш складні угруповання, утворення різного типу комплексів і т.п. Так, в концентрованих розчинах багатьох водорозчинних полімерів з гідрофобними групами, зокрема, похідних целюлози, триблоккополімерів, суміші полімерних поверхнево-активних речовин (ПАР), олігомерних ПАР і, особливо, амфіфільних графт-кополімерів, завдяки неелектричним взаємодіям відбувається зміцнення фізичної сітки міжмолекулярних зачіплювань та суттєвий ріст в’язкості.
Кількісне врахування усіх факторів неможливе, але в теоретичні рівняння часто вводять поправки, які пропорційні іонній силі розчину з емпіричним коефіцієнтом, що залежить від природи електроліту та розчинника.
Хімічні реакції між макромолекулами та поліелектролітні комплекси.
Поліелектроліти є реакційно здатними речовинами. Але хімічні реакції їх макромолекул мають певні особливості:
- реакції проходять завдяки кооперативним взаємодіям [ лат. cooperatio співробітництво]. При взаємодії двох макромолекул різних поліелектролітів зв’язки, які утворюються між ними, не є випадковими, а послідовними;
- в реакцію вступають лише кооперативні системи. Стан кожної з складових кооперативної системи залежить від стану сусідньої складової;
- системи повинні бути комплементарними [ лат.complementum засіб поповнення], тобто кооперативними системами здатними до кооперативних взаємодій.
Важливою групою реакцій, в які вступають поліелектроліти, є реакції між полікислотами та поліосновами, а також їх солями в водних розчинах, які призводять до утворення інтерполімерних солей (інтерполімерних комплексів).
Процес утворення полікомплексу і його властивості залежать від сили вихідних компонентів. Якщо вихідні поліелектроліти сильні і реакція між ними супроводжується значним тепловим ефектом, то процес утворення полікомплексу багатостадійний. Цей тип полікомплексів носить назву квазістабільних.

Безпосередньо після змішування розчинів різних за знаком поліелектролітів поліелектролітний комплекс утворюється аморфним, нерівноважним і нерегулярним. З часом в цьому полікомплексу проходять релаксаційні процеси, що спричиняють внутрикомплексну перебудову його структури, і полікомплекс переходить в більш упорядкований стан. На цьому етапі відбувається утворення іонних зв’язків або виправлення дефектів і збільшується глибина перетворення.
Вторинні полікомплекси завдяки гідрофобним взаємодіям можуть утворювати надмолекулярні структури типу фібрил або трьохмірну сітку. Схема утворення та перебудови інтерполімерного комплексу наведена на рис.60.
В розведених розчинах полікомплекс, як правило, виділяється в окрему високодисперсну фазу. Фізичний стан цієї фази залежить від густини заряду поліелектролітів, що реагують. При великій густині заряду на макромолекулах реагуючих поліелектролітів полікомплекси виділяються з розчину у вигляді драглю або високодисперсного осаду, який містить малу кількість розчинника.
При малій густині заряду (молекулярна маса поліелектроліту, що припадає на одну іонну групу не більше 300) утворюються коацерватні
полікомплекси, які виділяються в вигляді рідкої фази, що збагачена полімерними компонентами. Прикладом такого полікомплексу є продукт взаємодії гуміарабіку та желатину.
Сильні поліелектроліти, що здатні до взаємодії, можуть утворювати і стабільні полікомплекси. Стійкість цих комплексів визначають константи дисоціації вихідних поліелектролітів. Ці полікомплекси стійки практично в усьому інтервалі рН і руйнуються тільки в концентрованих розчинах низькомолекулярних електролітів у водно-органічних сумішах. Прикладом такого типу полікомплексів є сполука яку утворює полістиролсульфокислота з гідроксидом полівінілбензилтриметиламонію.
Полікомплекси з слабких поліелектролітів носять назву зворотньодисоційованих . Вони утворюються в кислому або лужному середовищі в реакціях нейтралізації або обміну. Полікомплекси цього типу стійкі тільки в обмеженому інтервалі рН, а утворення та руйнування цих комплексів відбувається кооперативно. До таких полікомплексів відноситься, зокрема, полі комплекс, який утворюють поліакрилова кислота та полівініламін :
в кислому середовищі (рН<7)
 ,
,
та в лужному середовищі (рН>7)
 .
.
Характерною особливістю таких кооперативних реакцій є витіснення сильної кислоти (HCl) або сильної основи (NaOH) внаслідок кооперативної взаємодії слабких полікислоти та поліоснови.
Кількісно оцінити вихід продуктів цих реакцій по кількості іонів Н+ та ОН- можна методом потенціометричного титрування, Тому, з кривих потенціометричного титрування сумішей полікислоти і солі поліоснови , а також поліоснови і солі полікислоти можна розрахувати ступінь перетворення (θ) в цих реакціях при любому значенні рН розчину
 ,
,
де СфГ – концентрація функціональних груп любого поліелектроліту, які можуть прийняти участь в утворенні сольових зв’язків.
Концентрацію сольових зв’язків (Ссз) , які утворюються по реакції , що проходить в кислому середовищі ( рН<7), можна розрахувати по формулі
 ,
,
де V0 – початковий об’єм системи, VNaOH – кількість титранту ( лугу ) в молях, що додана, [H+] - концентрація протонів в реакційній суміші, що утворилася за рахунок утворення полікомплексу, [H+]ПК – концентрація протонів, яку утворює полікислота при дисоціації. Остання є суттєвою, якщо
рНрозчину ≤ рНПК.
Концентрація сольових зв’язків, яка утворюється по реакції, що проходить в лужному середовищі ( рН>7 ), розраховується по формулі:
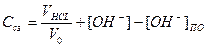 ,
,
де VHCL – кількість соляної кислоти, що додана,[OH-] - концентрація гідроксильних іонів в реакційній суміші, що утворилася за рахунок утворення полікомплексу, [ОH+]ПО – концентрація гідроксильних іонів, яку  утворює поліоснова при дисоціації. Остання є суттєвою, якщо
утворює поліоснова при дисоціації. Остання є суттєвою, якщо
рНрозчину ≥ рНПО.
Залежність θ-рН носить назву профілю реакції. Така залежність для системи поліакрилова кислота – полівініламін наведена на рис.61.
Вигляд цієї залежності вказує на те, що реакція між поліелектролітами відбуваються в вузьких інтервалах рН. Крутизна кривих θ- рН різна для різних пар поліелектролітів так і для однієї пари поліелектролітів в кислому та лужному середовищах.
Утворення полікомплексу з слабких полікислоти та поліоснови відбувається внаслідок зарядки малодисоційованого компоненту на повністю іонізованому ланцюгу полімерної солі. Тому криві іонізації полімер-полімерного комплексу не будуть спостерігатися при високих і при низьких значеннях рН, оскільки в першій і другій області ні один з компонентів повністю не заряджений.

 Полікомплекси можуть утворюватися не тільки завдяки виникненню сольових зв’язків, але й через виникнення водневих зв’язків між функціональними групами макромолекул компонентів системи. Прикладом полікомплексу такого типу є полікомплекс, який утворює в кислому середовищі поліакрилова кислота з поліетиленоксидом. Послідовне утворення водневих зв’язків між макромолекулами призводить до гідрофобізації частинок полікомплексу і згортання їх в компактні клубки.
Полікомплекси можуть утворюватися не тільки завдяки виникненню сольових зв’язків, але й через виникнення водневих зв’язків між функціональними групами макромолекул компонентів системи. Прикладом полікомплексу такого типу є полікомплекс, який утворює в кислому середовищі поліакрилова кислота з поліетиленоксидом. Послідовне утворення водневих зв’язків між макромолекулами призводить до гідрофобізації частинок полікомплексу і згортання їх в компактні клубки.
Серед методів встановлення комплексоутворення важливе місце посідає віскозиметрія. Наявність чи відсутність полікомплексу в водному розчині встановлюється по залежностям ηпит суміші/Σηі ─ склад системи.( Σηі – питома в’язкість компонентів ), рис.62.
Якщо в системі відсутня взаємодія компонентів, то
ηпит суміші =Σηі = 1, тобто ηпит суміші є адитивною величиною суми питомих в’язкостей розчинів вихідних полімерів. В умовах взаємодії компонентів системи має місце відхилення від адитивності і ηпит суміші ≠Σηі ≠1, рис.62 криві 2,3.
 Позитивне відхилення від адитивності є наслідком взаємодії поліелектролітів і утворення в розчині частинок, які за розміром більші за об’єми індивідуальних макромолекул компонентів системи, рис.62 крива 2.
Позитивне відхилення від адитивності є наслідком взаємодії поліелектролітів і утворення в розчині частинок, які за розміром більші за об’єми індивідуальних макромолекул компонентів системи, рис.62 крива 2.
Від’ємне відхилення від адитивності вказує на утворенні в розчині внаслідок взаємодії
його компонентів дуже щільних частинок полікомплексів рис.62 крива 3. Прикладом такої залежності є полікомплекс, який утворюється при взаємодії желатину (Ж) та гуміарабіку (ГА), рис.63.
Для наведеного вище прикладу взаємодії по водневим зв’язкам поліакрилової кислоти з поліетиленоксидом поява полікомплексу відображується на залежності характеристичної в’язкості розчину від його складу, що наведена на рис. 64. З рисунку видно, що при співвідношенні компонентів системи 1:1 відбувається найбільш щільне упакування макромолекул і утворюється полікомплекс.
Полікомплекси, які виділяються з розчину у вигляді нової фази, мають , як правило, стехіометричний склад (еквімолярні полікомплекси). Часто вони утворюються при співвідношенні компонентів 1:1. Якщо склад полікомплексу є нестехіометричним ( нееквімолярний полікомплекс), то такий полікомплекс є водорозчинним.
 Для водорозчинних полікомплексів вводяться поняття базового (БПЕ), або опорного (ОПЕ) поліелектроліту , та ліофілізуючого поліелектроліту (ЛПЕ). Ліофілізуючим поліелектролітом виступає той, якого в суміші більша кількість.
Для водорозчинних полікомплексів вводяться поняття базового (БПЕ), або опорного (ОПЕ) поліелектроліту , та ліофілізуючого поліелектроліту (ЛПЕ). Ліофілізуючим поліелектролітом виступає той, якого в суміші більша кількість.
Утворення водорозчинного полікомплексу відбувається постадійно. На першій стадії утворюється гідрофобний полікомплекс стехіометричного складу через утворення сольових зв’язків між макромолекулами з різним по знаку зарядом. Гідрофобізація виникає через пригнічення дисоціації функціональних груп макромолекул поліелектролітів, що вступили у взаємодію.
На другій стадії відбувається адсорбція надлишкових, вільних від взаємодії макромолекул ЛПЕ на частинках полікомплексу і, завдяки дисоціації їх функціональних груп у воді, ці частинки набувають можливості переходити в середовище розчинника ( воду).
Нестехіометричні комплекси можуть виникати і іншим шляхом. Схеми реакції, що призводять до їх утворення такі:
 а) полікомплекс може утворюватися через явище переносу, коли на первинний полікомплекс, який утворений полікислотою (ПК) і поліосновою (ПО), діє, наприклад, інша полікислота (ПК’), яка володіє сильнішими кооперативними властивостями. В цьому випадку полікислота, яка слабкіша, частково витісняється з полікомплексу цією сильною полікислотою по схемі:
а) полікомплекс може утворюватися через явище переносу, коли на первинний полікомплекс, який утворений полікислотою (ПК) і поліосновою (ПО), діє, наприклад, інша полікислота (ПК’), яка володіє сильнішими кооперативними властивостями. В цьому випадку полікислота, яка слабкіша, частково витісняється з полікомплексу цією сильною полікислотою по схемі:
б) порушення стехіометричності можливе при додаванні до полікомплексу надлишку одного з поліелектролітів, які взаємодіють. Цей надлишок забирає на себе частину протилежно зарядженого поліелектроліту, який до цього введення був зв’язаний в первинному полікомплексі. Внаслідок перерозподілу цього поліелектроліту утворюються полікомплекси з іншім співвідношенням компонентів по схемі:

в) порушення стехіометричності можливе і тоді, коли полікомплекс утворений одним з поліелектролітів, що має значну довжину ланцюгів макромолекул, а іншій має короткі ланцюги. Введення в такий полікомплекс
додатково полікомплексу, який теж має короткі ланцюги в другому поліелектроліті, але іншої хімічної будови, призведе до часткового перерозподілу коротких ланцюгів в обох полікомплексах по схемі:

г) нестехіометричний комплекс може утворюватися при частковій заміні коротких ланцюгів ліофілізуючого поліелектроліту на більш довгі. В цьому випадку короткі за розміром макромолекули ліофілізуючого поліелектроліту витісняються в розчин по схемі:

Електрохімічні властивості твердоподібних систем поліелектроліт – розчинник.
 Твердоподібні, у вигляді драглю, системи поліелектроліт – розчинник відрізняє те, що макромолекули в об’ємі утворюють просторову сітку. Функціональні групи ділянок макромолекул між зшивками (субланцюгів) у воді дисоціюють. Утворюються заряджені ланки і протилежно заряджені низькомолекулярні іони (проти іони),рис.65.
Твердоподібні, у вигляді драглю, системи поліелектроліт – розчинник відрізняє те, що макромолекули в об’ємі утворюють просторову сітку. Функціональні групи ділянок макромолекул між зшивками (субланцюгів) у воді дисоціюють. Утворюються заряджені ланки і протилежно заряджені низькомолекулярні іони (проти іони),рис.65.
Протиіони відокремлюються від полімерного ланцюга і вільно переміщуються всередині об’єму сітки, але вони не можуть виходити за її межі, оскільки в такому випадку порушується макроскопічна електронейтральність системи і виникають значні електричні сили взаємодії між зарядами сітки та протиіонами, що опинилися поза сіткою. Тому можна вважати, що поверхня драглю, що обмежує його об’єм, непроникна для протиіонів, а самі протиіони внаслідок свого переміщення в об’ємі сітки чинять на цю поверхню тиск. Це веде до додаткового всебічного розтягування зразка сітки , оскільки первісне розтягування сітки відбувається за рахунок взаємодії макромолекул сітки з гарним розчинником.
При погіршенні якості розчинника субланцюги скорочуються і , відповідно, зменшуються розміри зразка сітки. Коли в кожному субланцюгу відбувається перехід клубок – глобула зразок сітки різко зменшує свій об’єм. Це явище носить назву колапсу полімерної сітки.
Наявність заряду на субланцюгах і протиіонів в об’ємі сітки змінює картину колапсу сітки. Цю зміну можна проаналізувати з залежності вільної енергії полімерної сітки від коефіцієнту її набухання.

Вільна енергія субланцюга F(a) складається з суми енергетичного U (a) та ентропійного S (a) внесків (рівняння 12), де під а в даному випадку слід розуміти коефіцієнт набухання зразка полімерної сітки. Залежності U (a) та
S (a), що побудовані згідно рівнянь 17 та 20, а також для їх суми F(a) для незарядженої полімерної сітки наведені на рис.66а.
Поява заряду на субланцюгах мало змінює енергію U(a) ,оскільки додатковий вклад в неї кулонівської взаємодії при невеликій кількості заряджених ланок несуттєвий. Але ентропійний внесок в вільну енергію субланцюга значно доповнюється внаслідок ефекту всебічного розтягуванні сітки завдяки осмотичному тиску, що створюють протиіони, рис.66б. Внаслідок цього залежність F(a) приймає вигляд, що наведений на рис.66 в.
На цій залежності відображуються два локальних мінімуму вільної енергії.
Тепер, якщо згідно залежності для F(a), що наведена для незарядженої сітки (рис. 66а), одному мінімуму відповідає колапс сітки, який відбувається безперервно, то згідно залежності, що наведена на рис.66в колапс сітки повинен відбутися стрибкоподібно.
 На рис. 67 зображений колапс зарядженої внаслідок гідролізу сітки поліакриламіду у суміші вода - ацетон від процентного вмісту ацетону. Вода є гарним розчинником поліакриламіду, а ацетон –поганим. Видно, що при додаванні певної кількості ацетону (42%) до води, відбувається стрибкоподібний колапс сітки поліакриламіду, в ході якого об’єм сітки значно зменшується, приблизно в 20 разів.
На рис. 67 зображений колапс зарядженої внаслідок гідролізу сітки поліакриламіду у суміші вода - ацетон від процентного вмісту ацетону. Вода є гарним розчинником поліакриламіду, а ацетон –поганим. Видно, що при додаванні певної кількості ацетону (42%) до води, відбувається стрибкоподібний колапс сітки поліакриламіду, в ході якого об’єм сітки значно зменшується, приблизно в 20 разів.
Слід зазначити, що величина стрибку визначається кількістю функціональних груп субланцюгів, які продисоціювали. Чим більша кількість ланок стає зарядженими і ,відповідно, чим більший осмотичний тиск , що створюють протиіони, тим більша величина стрибку.
Якщо полімер сітки не несе заряду, але при зміні якості розчинника або зменшенні температури від