|
|
Що представляє з себе атомно-абсорбційний метод аналізу.
Атомно-абсорбційний аналіз (атомно-абсорбц. спектрометрія), метод кількісного елементного аналізу по атомних спектрах поглинання (абсорбції). Через шар атомних парів проби, одержуваних за допомогою атомізатора, пропускають випромінювання в діапазоні 190-850 нм. У результаті поглинання квантів світла атоми переходять у збуджені енергетичні стани. Цим переходам в атомних спектрах відповідають резонансні лінії, характерні для даного елемента. Відповідно до закону Бугера-Ламберта-Бера, мірою концентрації елемента служить оптична щільність A = lg (I0 / I), де I0 і I-інтенсивності випромінювання від джерела відповідно до і після проходження через поглинаючий шар.
Прилади для атомно-абсорбційного аналізу - атомно-абсорбційні спектрометри - прецизійні високоавтоматизовані пристрої, що забезпечують відтворюваність умов вимірювань, автоматичне введення проб і реєстрацію результатів вимірювання. В деякі моделі вбудовані мікроЕОМ. В якості прикладу на рис. наведена схема одного з спектрометрів. Джерелом лінійного випромінювання в спектрометрах найчастіше служать одноелементні лампи з порожнистим катодом, що заповнюються неоном. Для визначення деяких легколетючих елементів (Cd, Zn, Se, Ті й ін.) зручніше користуватися високочастотними безелектродними лампами.
Переклад аналізованого об'єкта в атомізований стан і формування поглинаючого шару пара певної і відтворної форми здійснюється в атомізатор - зазвичай в полум'ї або трубчастої печі. Найбільш часто використовують полум'я сумішей ацетилену з повітрям (макс. т-ра 2000 ° С) і ацетилену з N2O (2700 ° С). Пальник з щілиноподібні соплом довжиною 50-100 мм і шириною 0,5-0,8 мм встановлюють уздовж оптичної осі приладу для збільшення довжини поглинаючого шару.
Трубчасті печі опору виготовляють найчастіше з щільних сортів графіту. Для виключення дифузії пари через стінки і збільшення довговічності графітові трубки покривають шаром газонепроникного піровуглеводу. Максимальна температура нагріву досягає 3000 ° С. Менш поширені тонкостінні трубчасті печі з тугоплавких металів (W, Та, Мо), кварцу з ніхромовим нагрівачем. Для захисту графітових і металевих печей від обгорання на повітрі їх поміщають в напівгерметичні або герметичні камери, через які продувають інертний газ (Аr, N2).
Введення проб у поглинаючу зону полум'я або печі здійснюють різними прийомами. Розчини розпорошують (зазвичай в полум'я) за допомогою пневматичних розпилювачів, рідше - ультразвукових. Перші простіше і стабільніші в роботі, хоча поступаються останнім в ступені дисперсності утворюється аерозолю. Лише 5-15% найбільш дрібних крапель аерозолю надходить у полум'я, а інша частина відсівається у змішувальній камері і виводиться у стік. Максимальна концентрація твердої речовини в розчині зазвичай не перевищує 1%. В іншому випадку відбувається інтенсивне відкладення солей в соплі пальника.
Термічне випаровування сухих залишків розчинів - основний спосіб введення проб в трубчасті печі. При цьому найчастіше проби випаровують з внутрішньої поверхні печі; розчин проби (обсягом 5-50 мкл) вводять за допомогою мікропіпетки через дозувальне отвір в стінці трубки і висушують при 100 ° С. Однак проби випаровуються зі стінок при безперервному зростанні температури поглинаючого шару, що обумовлює нестабільність результатів. Щоб забезпечити сталість температури печі в момент випаровування, пробу вводять в попередньо нагріту піч, використовуючи вугільний електрод (графітову кювету) графітовий тигель (піч Вудріффа), металевий або графітовий зонд. Пробу можна випаровувати з платформи (графітового коритця), яку встановлюють у центрі печі під дозувальним отвором. У результаті значного відставання температури платформи від температури печі, що нагрівається зі швидкістю 2000 До / с, випаровування відбувається при досягненні піччю практично постійної температури.
Для введення в полум'я твердих речовин або сухих залишків розчинів використовують стрижні, нитки, човники, тиглі з графіту або тугоплавких металів, що поміщаються нижче оптичної осі приладу, так що пари проби надходять в поглинаючу зону з потоком газів полум'я. Графітові випарники в ряді випадків додатково підігрівають електричним струмом. Для виключення хутро. втрат порошкоподібних проб у процесі нагрівання застосовуються випарники типу циліндричних капсул, виготовлені з пористих сортів графіту.
Іноді розчини проб піддають в реакційному посудині обробці у присутності. відновників, найчастіше NaBH4. При цьому Hg, напр., Відганяється в елементному вигляді, As, Sb, Bi у вигляді гідридів, які вносяться до атомізатор потоком інертного газу. Для монохроматізаціі випромінювання використовують призми або дифракційні решітки; при цьому досягають дозволу від 0,04 до 0,4 нм. При атомно-абсорбційному аналізі необхідно виключити накладення випромінювання атомізатору на випромінювання джерела світла, врахувати можливу зміну яскравості останнього, спектральні перешкоди в атомізатор, викликані частковим розсіюванням і поглинанням світла твердими частинками і молекулами сторонніх компонентів проби. Для цього користуються різними прийомами, наприклад модулюють випромінювання джерела з частотою, на яку налаштовують приймально - реєструючий пристрій, застосовують двохвильову схему або оптичну схему з двома джерелами світла (з дискретним і безперервним спектрами). Найбільш ефективна схема, заснована на розщепленні і поляризації спектральних ліній в атомізаторі. У цьому випадку через поглинаючий шар пропускають світло, поляризоване перпендикулярно магнітному полю, що дозволяє врахувати неселективні спектральні перешкоди, досягають значень А = 2, при вимірюванні сигналів, які в сотні разів слабкіші.
Переваги атомно-абсорбційного аналізу - простота, висока селективність і малий вплив складу проби на результати аналізу. Обмеження методу - неможливість одночасного визначення декількох елементів при використанні лінійчатих джерел випромінювання і, як правило, необхідність переведення проб в розчин. Атомно-абсорбційний аналіз застосовують для визначення близько 70 елементів. Не визначають гази і деякі інші неметали, резонансні лінії яких лежать у вакуумній області спектру (довжина хвилі менше 190 нм). Із застосуванням графітової печі неможливо визначати Hf, Nb, Та, W і Zr, які утворюють з вуглецем важколетючі карбіди. Межі виявлення більшості елементів у розчинах при атомізації в полум'ї 1-100мкг / л, в графітової печі в 100-1000 разів нижча. Абсолютні межі виявлення в останньому випадку становлять 0,1-100 нг. Щодо стандартного відхилення в оптимальних умовах вимірів досягає 0,2-0,5% для полум'я і 0,5-1,0% для печі. В автоматичному режимі роботи полум'яний спектрометр дозволяє аналізувати до 500 проб на годину, а спектрометр з графітової піччю -до 30 проб. Методи атомно-абсорбційного аналізу застосовують також для вимірювання деяких фізичних і фіз.-хімічних величин - коефіцієнт дифузії атомів у газах, температур газового середовища, теплоти випаровування елементів і ін.
2. Явище люмінесценції, які види люмінесценції використовують у фізико – хімічних методах аналізу.
Люмінесценція – особливий вид світіння речовин без підвищення температури – відома ще з глибокої старовини. Термін "люмінесценція" і класифікацію типів світіння вперше запропонував німецький фізик Відеман. Однак його визначення було неповним. Під час фотолюмінесценції частка починає інтенсивно світитися в результаті захоплення квантів активуючого світла. Причому, повертаючись до вихідного стану, вона віддає отриману енергію у вигляді світла, довжина хвилі якого більша довжини хвилі джерела збудження.
Отже, люмінесценцією називають світіння атомів чи молекул, яке виникає в результаті електронного переходу в частинках речовини при їх переході із збудженого стану в не збуджений.
Класифікують явища люмінесценції за часом та методом збудження. За часом післясвітіння розрізняють два типи люмінесценції – флуоресценцію – світіння яке миттєво зникає після припинення дії джерела збудження і фосфоресценцію, світіння, продовжується певний проміжок часу.
 В залежності від методу збудження розрізняють фотолюмінесценцію – свічення, яке виникає при поглинанні світлової енергії; катодолюмінісценцію – основану на свіченні речовин при поглинанні катодних променів (електронів) та хемілюмінесценцію – свічення, яке виникає при протіканні хімічних реакцій.
В залежності від методу збудження розрізняють фотолюмінесценцію – свічення, яке виникає при поглинанні світлової енергії; катодолюмінісценцію – основану на свіченні речовин при поглинанні катодних променів (електронів) та хемілюмінесценцію – свічення, яке виникає при протіканні хімічних реакцій.
Всі люмінесціюючи речовини мають загальну назву – люмінофори. У найпростішому вигляді процес, збудження і свічення можна зобразити схемою, наведеною на рис. 1, з якого видно, що енергія випромінювання молекули завжди менша від енергії збудження.
Рис. 1. Збуджений та нормальний стани молекули та переходи між ними в процесі люмінесценції де Н – нормальний стан молекули із станами 0,1, 2, 3, 4, З – збуджений стан молекули із станами 0,1, 2, 3, 4.
На основі цього було встановлено, що спектр люмінесценції зміщений відносно спектру поглинання в сторону довших хвиль (закон Стокса-Ломмеля). Доведено також дзеркальну подібність спектрів поглинання і спектрів випромінювання люмінесценції для складних молекул. Дзеркальна симетрія спектрів родаміну в ацетоні наведена на рис. 2.

Рис. 2. Дзеркальна симетрія спектрів родаміну в ацетоні.
1 – спектр поглинання, 2 – спектр випромінювання.
Різницю між максимумом спектру поглинання і максимумом люмінесценції Х називають стоковим зміщенням. Чим більша величина зміщення для даної люмінесціюючої речовини, тим вища чутливість визначуваної речовини люмінесцентним методом.
Повнота перетворення енергії збудження при люмінесценції характеризується енергетичним виходом Ве, який являє собою відношення випромінюваної енергії люмінесценції Ел до поглинутої енергії збудження Ев.
Ве= Ел / Ев.
Повноту перетворення енергії можна охарактеризувати також величиною квантового виходу люмінесценції Вк, який дорівнює відношенню числа випромінюваних квантів при люмінесценції Nл до числа поглинутих квантів Nв при збудженні:
Вк= Nл / Nв.
Оскільки енергія кванта оптичного випромінювання рівна: Е = hν, то зв'язок між енергетичним та квантовим виходом люмінесценції можна виразити рівнянням:
Ве= Ел / Ев = Вк (λв/ λл)
Енергетичний вихід люмінесціюючого випромінювання залежить від довжини хвилі збудженого світла (закон Вавилова). Графічно ця залежність показана на рис. 3.
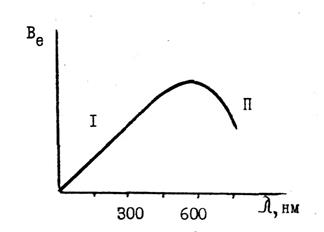
На ділянці 1 кривої величина енергетичного виходу росте пропорційно довжині хвилі збудженого світла; далі в ділянці накладання спектрів поглинання і випромінювання відбувається різке падіння виходу (ділянка ІІ).
Рис. 3. Залежність виходу випромінювання від довжини хвилі збуджуючого світла.
Для ділянки кривої 1 можна записати: Ве=аλв, де а - коефіцієнт пропорційності.
Але оскільки Ве= Ел / Ев = Вк (λв/ λл), то, об'єднавши два рівняння і знаючи, що речовина при люмінесценції випромінює світло певної довжини хвилі, одержимо вираз:
Вк = аλк = const,
з якого випливає, що квантовий вихід люмінесценції залишається сталим на ділянці 1 наведеної кривої при збільшенні довжини хвилі збудженого світла до 500 – 600 нм. Саме ділянку спектру 1 (від 100 до 600 нм) використовують для кількісного визначення речовин. На практиці для проведення люмінесцентного аналізу багатьох речовин використовують ультрафіолетові промені світла з більшою енергією кванту, які одержують в основному за допомогою ртутних ламп.
3. Задача.
При аналізі на хром по методу трьох еталонів на мікрофотометрі МФ -2. Вивірено почорніння (S) ліній гомологічної пари в спектрах еталонів та досліджувального зразка. Знайти відсотковий вміст хрому (Сr) по дослідним даним:
Еталон І ІІ ІІІ
Сср 0,50 1,23 4,87
S 0,27 0,23 0,27
SCr 0,07 0,37 0,86
Аналізуючий зразок має SCr = 0,61 і STl>0,25.
Розв‘язування:
В методі трьох еталонів використовується залежність різниці ( S) поглинання ліній гомологічної пари від логарифму концентрації дослідного елементу при визначених умовах ця залежність близька до лінійної.
За показниками вимірної шкали мікрофотометра знаходимо
∆S (∆S2-Sсп-SFe ), для трьох еталонів: ∆S2=0,37-0,23=0,14; ∆S3=0,86-0,27=0,59. Визначаємо за даними таблиці логарифми концентрацій:
lg С1=-0,30; lg С2=0,09; lg С3=0,62, та будуємо калібровочний графік у координатах ∆S - lg С (рис. 1). Знаходимо ∆S для аналізую чого зразка:
∆Sх=0,61-0,25=0,36. Ссп=0,24%.
ЕТАЛОННА ВІДПОВІДЬ
Варіант №7
1. Прилади та речовини для люмінесцентного аналізу.
Метод люмінесцентного аналізу в медицині став більш успішно розроблятися з розвитком вивчення вторинної люмінесценції. Остання представляє світіння, що виникає після зафарбування тканин спеціальними барвниками – фото люмінофорами. Органічні фотолюмінофори, що випромінюють під дією ультрафіолетових чи променів видимої частини спектра, часто називають флуоресцентними барвниками, чи флуорохромами.
Флуоресцеїн (диоксифлуоран) (C20H12O5) – органічна сполука, барвник трифенілметанової групи, червоний кристалічний порошок, розчиняється в спирті, ефірі та водних лугах. Назва "флуоресцеїн" дано сполуці тому, що в лужних розчинах він має сильну флуоресценцію (у концентраціях до 1 : 2000000).
Отримують флуоресцеїн при нагріванні фталевого ангідриду з резорцином. У лужному середовищі оптимальна концентрація його 0,8 г/л, колір флуоресценції жовто-зелений, відносна яскравість 0,26 нт.
Флуоресцеїн майже нерозчинний у воді, тому для введення цього препарату застосовують його натрієву сіль. Це один з найяскравіших флуорохромів.
Стрімкий розвиток молекулярної біології та біотехнології потребує швидких і зручних методів детекції нуклеїнових кислот (НК) та білків. На заміну радіоактивним міткам, що традиційно застосовуються з цією метою, приходять ефективніші, зручніші, дешевші та безпечніші спектрально-люмінесцентні методи детекції.
Апаратура для люмінесцентних досліджень повинна бути портативною, зручною, забезпечувати проведення діагностичних спостережень у будь-яких умовах, мати оптико-світлотехнічну систему для концентрації випромінювання на визначену ділянку. Основними вузлами апаратури для люмінесцентного аналізу є освітлювач із світлофільтрами, кювети, діафрагми і пристрій для вимірювання інтенсивності свічення. Освітлювачем для люмінесцентного аналізу, як правило, використовують ртутні лампи. Приймачем виступає фотоелемент або фото помножувач. Принципова схема лабораторного флуорометра ЄФ-ЗМ, призначеного для кількісного аналізу вітамінів та інших люмінесціюючих речовин, показана на рис. 4.

Рис. 4. Схема лабораторного флуорометра ЄФ-3М
1.кварцова лампа;
2.діафрагма;
3.заслінка;
4.фільтр;
5.кварцова оптика;
6.посудина з досліджуваним розчином;
7.кварцова оптика;
8.світлофільтри;
9.фотоелементи.
Світло від кварцової лампи 1, проходячи через діафрагму 2, світлофільтр 4 і кварцову оптику 5, потрапляє на посудину з досліджуваним розчином. Люмінесцентне свічення досліджуваного розчину проходить через кварцову оптику 7, вторинні світлофільтри 8, потрапляє на фотоелемент 9. Фотоелемент, перетворюючи світлову енергію в електричну, подає її на електронний підсилювач, в анодний ланцюг якого підключений мікроамперметр. Покази мікроамперметра прямо пропорційні концентрації люмінесціюючої речовини.
Спочатку результати люмінесцентних досліджень оцінювалися лише візуально і за допомогою фотографування.
У практичній роботі користалися візуальним лабораторним фотометром моделі ВФМ-57. Прилад дозволяє проводити вимірювання в області малих яскравостей, причому як білих, так і кольорових поверхонь. Межі виміру малих яскравостей приладу від 5-10 до -0,00003 нт. Верхня межа виміру високих яскравостей до 1 · 106 нт у "кольоровому" світлі і до 5 · 106 нт у "білому" світлі.
Принцип дії приладу заснований на візуальному порівнянні яскравості двох фотометричних полів, з яких одне - вимірюване, інше - поле порівняння. Для реєстрації яскравості кольорових поверхонь до приладу додається набір з дев'яти кольорових світлофільтрів. Основні вузли фотометра малих яскравостей- вимірювальна голівка, стійка, яка живить пристрій.
Для фотоелектричного фотометрування сконструювали апаратуру для графічного запису процесу фотоелектричного фотометрування. Змонтована на базі ФЗУ-54 з режимом живлення 1800 В, вона дозволяє проводити дослідження на рівні мікроциркуляції і записувати у виді графічних кривих картину світіння, що спостерігається. Прилад складається з фіброволоконного світловоду, світлочутливого елементу (ФЭУ-54), джерела живлення й одноканального элекфокардиографа ЭКСПЧТ-4 чи самописи Н-390.
Аналізатори типу «Флюорат» є прикладом доступної лабораторної апаратури, що реалізує можливість фотолюмінісцентних та хемілюмінесцентних вимірювань. Застосовані в них імпульсні плазмові джерела світла забезпечують високу чутливість, широкий спектральний діапазон та можливість кінетичних вимірювань в часових інтервалах до 105-106 секунд. Крім того, ці прилади дозволяють легко реєструвати люмінесценцію при низьких температурах (77 К).
2. Яким рівнянням виражаються закон світло поглинання: Бугера-Ломберта-Бера.
Бер встановив коефіцієнт поглинання пропорціональний концентрації поглинаючої речовини:
К = а×С;
Е – коефіцієнт незалежний від концентрації;
С – концентрація речовини.
Закон Бугера-Ланберта-Бера розглядає поглинаючи світлового потоку рідиною концентрації при вимірюванні поверхні поглинаючого шару.
Закон Бера: Зміна поглинання світлового потоку шаром постійної товщини відбувається при вимірюванні концентрації.
Рівняння основного закону колориметрії:
Ів= І0×10-Сl.
Ів - інтенсивність світлового потоку;
І0 - інтенсивність падаючого світлового потоку;
L – товщина шару.
Якщо концентрація С в моль/л, а довжина l – 1 см, то Е – молекулярний коефіцієнт поглинання. Е – соnst, залежить від довжини падаючого світла.
Значення Е характеризує чутливість колориметричного визначення: чим більше значення Е, тим вища чутливість. Прозорість (пропускання) розраховується формулою:
Т =  ×10-ЕCl;
×10-ЕCl;
Оптична щільність (поглинання – екстензія Е).
Д=Е lg = lg = ЕСl;
3. Задача.
Для двох еталонних водних розчинів хлориду кальцію з вмістом хлориду кальцію  і
і  знайдемо значення показника переломлення, що дорівнює
знайдемо значення показника переломлення, що дорівнює 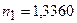 і
і  , та
, та  .
.
Розрахувати вміст хлориду кальцію (х), % в аналізуємо му розчині для:
а)  ;
;
Б) невідомий.
Рішення.
а) За формулою розраховуємо значення фактору (F):

 .
.
Вміст хлориду дорівнює:

 .
.
б) Користуючись інтерполяцією між п і х розраховуємо вміст хлориду кальцію:

 .
.
Висновок: Спосіб розрахунку застосовують при використанні даних з рефрактометричних таблиць. Вміст СаСl2 дорівнює 3,2%.
ЕТАЛОННА ВІДПОВІДЬ
Варіант №8
1. У чому полягає сутність амперметричного титрування.
Амперметричне титрування – різноманітність вольтамперометричного методу. Воно застосоване на вимірюванні величини струму між електродами електрохімічної ячійки, до яких прикладена деяка напруга, як функції об’єму прибавленого титрування. У відповідності з рівнянням Іцісовача ів=Кl, дифузійний струм в ячійці, концентрація такої речовини зменшується, або збільшується й дифузійній струм. Точку еквівалентності фіксують по-різному. Зміненню падіння, або зросту дифузійного струму, що відповідає закінченню реакції титрує мого розчину з титратом.
Основане на тому, що кінцеву точку титрування знаходять по зміні сили граничного дифузійного струму, який проходить через розчин при сталій напрузі між індикаторним електродом і електродом порівняння. За результати титрування будують графік залежності струму і об’єму робочого розчину на кривій знаходять точку перетину двох віток кривої, яка відповідає кінцевій точці титрування, тобто вимірюють об’єм розчину, який витратився на титрування і за основною формулою об’ємного аналізу проводять розрахунки.
2. Хвильова природа світла. Дати визначення дисперсії світла.
Дисперсія світла – залежність оптичних речовин, тобто його показника заломлення від довжини хвилі рухомого світла, а також явища у яких знаходиться ця залежність. Хвилі різної довжини розповсюджуються у якому-небудь середовищі з різними швидкостями і з різними заломленнями.
Так, як світлові промені різних кольорів мають різну довжину, то при пересуванні крізь скляну призму, вони відхиляються на окремі кути. Внаслідок дисперсії прозоре світло, що проходить крізь зйомки середовища, розкладаються на сім основних кольорів, що поступово переходять один в одного.
Таке світло називається монохроматичним.
3. Задача.
При аналізі алюмінієвого сплаву на кремній по методу одного еталону одержали затемнення (S) ліній гамологічної пари у спектрах еталону (Ssi=1,09 та Sal=0,37 при Сsi=0,95%) і аналізує мого зразку:
Ssi=0,86 та Sal=0,34
Визначити відсоткове одержання кремнію в зразку, якщо ∆S=0,
при Сsi=0,45 %.
Рішення.
У методі одного еталону калібрований графік у координатах ∆S=0, при lg Сsi= lg 0,45=-0,35.
За даними фотометрування знаходимо координати другої точки і будуємо калібрований графік: ∆S1=Ssi-Sal=1,09-0,37=0,72; lg l1= lg0,95=-0,02.
Визначаємо ∆Sх для досліджувального зразка: ∆Sх=0,86-0,34=0,52 і з допомогою графіка знаходимо зміст кремнію: lg Сх=-0,11, що відповідає Сsi=0,78 %.
ЕТАЛОННА ВІДПОВІДЬ
Варіант №9
1. Що називають оптичною щільністю і коефіцієнтом припускання.
Коефіцієнт екстензії називають коефіцієнтом світлопоглинання або коефіцієнтом поглинання. Це не зовсім точно тому, що так коефіцієнт екстензії і коефіцієнт поглинання – різні величини (Е=К/2,3).
Оптична щільність, коефіцієнти поглинання і коефіцієнти погашення залежать від природи поглинаючих речовин і розчинника. Довжина хвилі поглинаючого світла, температури.
У якісному аналізі при характеристиці даної речовини визначають хвилі max або min у спектрі поглинання.
Для цього необхідно записати на спектрографі УВІ, спектр поглинання цього аналізує мого розчину і дорівнюють одержаний спектр у відомий спектром з’єднання яке може бути у аналізуємому розчині. Звичайно цей спосіб використовують для підтвердження присутності відкриває мого компоненту, наприклад, для підтвердження дійсності фармакологічного активу речовин.
2. На чому заснований рефрактометричний аналіз.
Рефрактометричний метод заснований на вимірюванні показника світлопоглинання (показника приломлення) рідним або кристалів.
Абсолютний показник приломлення N – це відношення швидкості С0 розповсюджуються світло у вакуумі до швидкості з його розповсюдження у даній середі: N=С0/С.
Відносний показник приломлення n – це відношення швидкості розповсюдження світла у повітрі С повітря до швидкості розповсюдження у даній середі: n=Спов/С.
Показник приломлення залежить від природи приломлюючого світла, температури, концентрації рідини, від напрямку падаючого світла на кристал.
Звичайно на практиці показник приломлення визначають при довжині хвилі падаючого світла: l=589,3 мм, відповідаючи положенню ліній у спектрі вилучення натрію.
Зміни показника приломлення чистих рідин і розчинів проводять на спеціальних рефрактометрах.
Тугу рефрактометру Аббе (рідше рефрактометру Пульфрила).
3. Задача.
Розрахунки з використанням молярного коефіцієнту гашення.
Навіску вагою m=0,0300 г препарату ретоналацетату розчинниками у абсолютному етанолі й отримали V(х)=100 похідного аналізує мого розчину. Вибрали 1 мм цього розчину, прибавили до нього 99 мл абсолютного етанолу й отримали 100 мл виміряє мого розчину. Визначили оптичну щільність А(х) виміряє мого розчину на спектрофотометрі, при довжині хвилі l=326 нм в кюветі з товщиною поглинаючого шару l=1 см, рівною А(х) = 0,456.
Розрахувати зміст ретинолу ацетату у відсотках в 1 г препарату, якщо молярний коефіцієнт гашення спиртового розчину ацетату С23Н32О2 рівняється Е=50900 л×моль-1 см-1, при l=326 нм.
Розв’язання.
Відсотковий вміст х ретинолу ацетату в 1 г препарату рівно:
Х=  ·100 %,
·100 %,
де m(x) – вага (г) ретинолу ацетату у вихідному аналізуємому розчині (в навісці m=0,0300 г препарату), яка дорівнює: m(x)=С(х)·М(х)×V(х), де С(х) – концентрація ретинолу ацетату у вихідному аналізуємому розчині, моль/л.
М(х)=328,50 – молярна вага ретинолу ацетату;
V(х)=100 мл=0,1 л – об’єм вихідного аналізуємого розчину.
Для визначення С(х) розрахуємо спочатку концентрацію С вимірює мого розчину. У відповідності з основним законом світлопоглинання А=Еcl концентрація С дорівнює.
С=А/Еcl=0?456/50,900·1=8,9·10-6, моль/л
Концентрація С(х) вихідного аналізує мого розчину в 100 разів більше концентрації вимірювального розчину:
С(х)=100С=100·8,9·10-6=8,9×10-4, моль/л
Зараз знаходимо М(х):
m(х)=8,9·10-4·328,50·0,1=0,0292 г, відрахуємо х: Х=  ·100 % =97,3 %.
·100 % =97,3 %.
ЕТАЛОННА ВІДПОВІДЬ
Варіант №10
1. Як знайти концентрацію визначаємої речовини при диференціальній фотометрії.
Готують ряд (п’ять – десять еталонних розчинів визначаємого розчину з різноманітною точно заданою концентрацією С0, С1, С2...Сn.
Спочатку при вибраній довжині хвилі в обидва канали спектрофотометра, поміщують однакові кювети з одним і тим же еталонним розчином (концентрація визначаємої речовини дорівнює С0) відносно якого будуть проводити послідуючі заміри й установлюють шкалу оптичної щільності в положенні А=0.
Потім при цій же постійній аналітичній довжині хвилі замірюють оптичну щільність Аі (і=1,2...n) кожного еталонного розчину й оптичну щільність Ах аналізує мого розчину водного еталонного розчину з концентрацією С0 й особистою оптичною щільністю А0 (відносно чистого розчинника) після чого знаходять концентрацію Сх визначаємої речовини в аналізуємому розчинні наступним способом.
Розрахунковий спосіб. При цьому способі передбачається виконуваність основного закону світлопоглинання. У відповідності з цим законом можливо написати:
Ах=Еl(Сх-С0);
Сх=С0=Ах/Еl,
Сх=С0+Ах/Еl,
де Е – молярний коефіцієнт гасіння визначаємої речовини,
l – товща поглинаючого шару;
Якщо ввести фактор переліку F=1/Еl, то останнє рівняння можна переписати у вигляді: Сх=С0+FАх.
Ця речовина й використовується для розрахунку концентрацій Сх визначаємої речовини на основі вимірювання Ах, а при відомій концентрації С0 еталонного розчину зрівняння.
2. Привести рівняння Нернста і пояснити сенс вхідних величин.
Рівняння було (в 1890 р.) вперше виведене В.Нернстом. У розбавлених розчинах замість активності можна підставляти концентрації і рівняння прийме вигляд:
Е=Е0+  InCm2+
InCm2+
Таким чином Е0 – електродний потенціал, який виникає при занурені металу в розчин, в якому активність іонів металу рівна 1 моль/л.
Ця величина отримала назву стандартного електродного потенціалу.
3. Задача.
Питомий коефіцієнт поглинання водного сіркокисневого розчину дихромату калію K2Cr2O7, при довжині хвилі l=455 км=Е=61.
Розрахувати молекулярний коефіцієнт поглинання Е дихромату калію в том розчині.
Рішення. В відповідності з множниками раніше Е=µ×Е/моль, де.µ=294,185 молекулярна маса дихромату калію.
Послідовно Е=294,185·  »1795 л · моль-1.
»1795 л · моль-1.
ЕТАЛОННА ВІДПОВІДЬ
Варіант №11
1. На чому заснований кондуктометричний метод. Від чого залежить питома електропровідність.
Кондуктометричний аналіз ґрунтується на використанні залежності від електропровідності. Від електропровідності розчинів електролітів-провідників другого роду судять на основі вимірювання їх електричного опору в електрохімічній ячійці, яка представляє скляний посуд 2 мл, вливши в нього електроди між якими находяться дослід жувальний розчин електроліту. По фізичному суміш титану електропровідність, це електрична провідність шару електроліту, який знаходиться між сторін куба з довжиною сторін 1 см. Питома електропровідність залежить від природи електроліту і розчинювала від концентрації розчину, від температури.
З збільшенням концентрації розчину електроліту його питома електропровідність спочатку зростає, потім проходить через максимум, після чого зменшується. В розчинах слабких електролітів з ростом концентрації поліпшується ступень дисоціації молекул електроліту, що призводить до зменшення числа іонів.
Питома електропровідність розчинів електролітів зменшується з ростом температури, внаслідок зниження в’язкості розчинів, що призводить до підвищення швидкості руху іонів. Таким чином, якщо питома електропровідність характеризується електричною провідністю одиничного об’єму розчина електроліту.
2. Описати принцип кулонометричного аналізу.
Кулонометричний аналіз ґрунтується на використанні залежності між масою (m), речовини яка прореагувала, при електролізі в електрохімічній ячійці, і якістю струму Q, проходження через електрохімічну ячійку, при електролізі тільки речовини. Відповідності з законом електролізу М. Фарадея маса зв’язана з кількістю струму Q.
Якщо відома кількість струму Q, то можливо розрахувати масу. То справедливо в том випадку, як кількість струму Q проходження через електрохімічну ячійку. Використано тільки на електроліз даної речовини.
Оскільки колунометричний аналіз проводять в амперметричному розпорядку тобто, при постійному електричному струмі; = const, чи при контрольованому постійному потенціалі робітничого електрода, коли електричний струм вимірюється в процесі електролізу.
Дві однакові пробірки діаметром 2--2,5 см і висотою 25--30 см вставляють у штатив, в обидві пробірки наливають реактиви. в першу -- досліджуваний розчин, а в другу поступово добавляють стандартний (з відомою концентрацією речовини) розчин із бюретки. Стандартний розчин добавляють доти, поки інтенсивність забарвлення обох розчинів не зрівняється при однакових об'ємах. Розчини в обох пробірках весь час перемішують. Вміст речовини знаходять за об'ємом добавленого стандартного розчину. Це легко зробити, тому що концентрація стандартного розчину відома. Перевага методу колориметричного титрування перед методом шкали в тому, що цей метод можна застосувати тоді, коли забарвлена сполука недостатньо стійка в часі (тіоціанат феруму).
У колориметрах занурення зрівнюють інтенсивність забарвлення, змінюючи товщину шару розчину. Досліджуваний і стандартний розчини наливають у циліндричні скляні посудини, які за допомогою спеціальних механізмів можуть опускатись і підніматись. У ці циліндри вільно входять нерухомо закріплені скляні палички з оптичного скла. При опусканні чи підніманні циліндрів змінюється товщина шару забарвленого розчину, що фіксується на спеціальних шкалах, з'єднаних через покажчик рівня розчину з циліндрами. Однакова товщина шару в обох циліндрах при однаковій інтенсивності забарвлення обох половин поля зору свідчить про однаковість концентрацій обох розчинів. Однаковості поля зору можна досягти, змінюючи товщину шару одного з розчинів. Концентрацію досліджуваного розчину обчислюють за формулою:
Cx = Cст*lст/lx
lx - lст - товщина шару розчину.
3. Задача.
При визначенні олова у бронзі методом постійного графіку сфотографували на одній пластинці спектри чотирьох еталонів і отримали слідуючи результати.
Еталон І ІІ ІІІ ІV
S=Ssn-Su 0,690 0,772 0,831 0,910
Ssn, % 6,23 8,02 9,34 11,63
Спектр одного з елементів знятий через трьохступінчатий послаблювач, при цьому для обраної лінії Sn різність почорнінь двох ступенів ∆Sступ=1,065.
Спектр аналізує мого зразку зняли на др. Пластинці також через трьохступінчатий послаблювач, отримали слідуючи результати і
∆Sступ=0,925 – різність почорнінь двох ступенів обраної лінії олова ∆Sступ =0,695 – різність почорнінь ліній літологічної пари Sn-Cu. Визначити % вміст в зразку .
Рішення. Знаходимо по значенню Сsn логарифми для чотирьох еталонів
(lg C1=0,795; lg C2=0,904; lg C3=0,970; lg Cn=1,038) і будуємо калібровочний графік у координатах. ∆S-lg C.
Визначаємо перевідний помножувач h, який дозволяє використовувати цей графік для аналізу зразків, спектри яких отриманні на різних фотопластинках:
K=  =
=  ;
;
де  і
і  - коефіцієнт контрастності обох фотопластинок зі спектром еталонів і аналізуємого зразка.
- коефіцієнт контрастності обох фотопластинок зі спектром еталонів і аналізуємого зразка.
Підставляємо численні значення ∆S: к=  =1,151.
=1,151.
Знаходимо значення різності положень лінії гомологічної пари у спектрі аналізує мого зразку з розрахунком перевідного положення:
∆Sx=k·∆Sx=1,151=0,695=0,800
По каліброваному графіку знаходимо lg Cx=0,933, що відповідає СSn=8,57 %.
Перевідний компонсувач K , користуючись трьохступінчатим послаблювачем. В цьому випадку необхідно знати різність почорнінь обох з’єднань ліній основи на основній пластинці, на якій знятий спектр аналізую чого зразку. Відсіля К=∆Sосн/∆Sосн.
ЕТАЛОННА ВІДПОВІДЬ
Варіант №12
1. Написати сутність хроматографічного аналізу.
Хроматографія – це область науки, вивчаюча процеси, основані на переміщенні зони речовини здовж шару сорбенту у катіоні і зв’язані з многократним повторенням сорбційних і десорбцій них актів.
Класифікація по механізму розподілення речовини:
а) адсорбційна хроматографія – заснована на використанні неоднакової здібності розподілених компонентів вступати в специфічну взаємодію з поверхнею адсорбенту – за рахунок адсорбції;
б) розподілення хроматографії – заснована на використанні різниць в коефіцієнтних розподіленнях розділених компонентів являючи собою рідину;
в) іонообмінна хроматографія – заснована на використанні різної здібності іонів, розподіляючих компонентів, які знаходяться в РФ (рідкий розчин) до обміну з іонами РФ).
г) хіміхроматографія – заснована на використанні різної здібності компонентів, розподіленої суміші вступати в ті або інші хімічні реакції з реагентами, які входять в склад РФ. При цьому розрізнюють такі види хіміхроматографії, як осадочна, біоспецифічна хроматографія;
д) ексклюзивна (проникаюча) хроматографія – заснована на використанні відмінності між розмірами (ефективними діаметрами) часто розподілених компонентів розмірами РФ, яка представляє собою сорбент – пориста речовина.
Класифікація по техніці експерименту розрізнюють: колоночну,
плоскостінну (тонку та паперову) хроматографію.
Колоночна хроматографія – для розподілення компонентів, використавши хроматографічні колонки , заповнені сорбентом.
Капілярна хроматографія – у якості хроматографічних колонок використовують капілярні трубки з скла або другого металу.
Паперова хроматографія – частіше усього використовують спеціальний хроматографічний напір, волокна якого покриті тонким шаром води або другої рідини.
2. На чому заснований принцип спектрофотометрії.
Метод спектрофотометрії заснований на використанні здатності речовин селективних, це коли суворо визначені довжини хвиль.
Принцип методу заклечається у наступному і луч світла від джерела збудження проходе через скляну або кварцову кювету зафіксованої товщі, заповнену аналізуємим розчином. Світло, яке прийняло через кювету з розчином, направляються на вхідну щіль спектрофотометру, у якому він розкладається на спектр.
Спектрофотометри забезпечують достатньо високу степінь мотохроматизацйії світла (~ 0,2-5 мм) за рахунок використання спирання елементів – призм і діфракціонних решіток. Після розложення в спектр електромагнітна енергія світла регіструється автоматично або по точкам до форми спектральної кривої, записуємої у вигляді графіка функції. В сучасних хімічних дослідженнях широко застосовують спектральні методи. Ці методи все більше застосовують в технічному аналізі хіміко-фармацевтичних препаратів, в аптечній практиці. Серед оптичних методів найбільш доступною, а тому і самою поширеною є видима і ультрафіолетова (УФ) спектрофотометрія, яка дозволяє відносно нескладному обладнанні швидко і точно проводити кількісний аналіз речовин.
Спектрофотометрія у видимій області і УФ-областях дозволяє оцінювати ступінь чистоти речовини, ідентифікувати по спектру різні сполуки, визначити константи дисоціації кислот і основ, досліджувати процеси комплексоноутворення.
Інфрачервоні (ІЧ) спектри дають характеристику речовин. Наявність в ІЧ-спектрах тих чи інших полос поглинання дозволяє розшифровувати структуру речовини.
УФ-спектрофотометричне вимірювання проводять в розчинах. Як розчинники використовують очищену воду, кислоти, луги, спирти (метанол, етанол), деякі інші органічні розчинники. Розчинник не повинен поглинати в тій чи іншій області спектра, що і аналізуємо речовина. Характер спектра (структура і положення полос поглинання) може змінюватися в різних розчинниках, а також при зміні рН середовища.
Методом УФ-спектрофотометрії використовують для визначення ідентичності, чистоти і кількісного вмісту лікарських препаратів.
Вивчення спектрів поглинання хімічних речовин з різною структурою дало можливість установити, що основними факторами, які обумовлюють поглинання світла, є наявність так званих хромофорів, тобто ненасиченість (подвійні чи потрійні зв’язки), наявність карбонільної, карбоксильної, амідної, азо-, нітрозо-, нітро- та інших функціональних груп. Кожна функціональна група характеризується поглинанням в певній області спектра. Але є ряд факторів (присутність декількох хромофорних груп, вплив розчинника та ін.) приводять до зміщення смуг поглинання в сторону більших довжин хвиль (батохромне зміщення) або в сторону коротких довжин хвиль (гіпсохромне зміщення). Крім зміщення може спостерігатися ефект збільшення (гіперхромний) чи зменшення (гіпохромний) інтенсивності поглинання.
В зв’язку з цим для ідентифікації речовин по її УФ0спектру застосовують метод порівняння із спектром відомої речовини, одержаний в тих же умовах. Характеристикою спектра поглинання речовини є положення максимумів (мінімумів) поглинання, а також інтенсивність поглинання, що характеризується величиною густини чи питомого показника поглинання при даній довжині хвилі.
Інфрачервоні (коливальні) спектри використовуються для ідентифікації лікарських препаратів ІЧ-спектри більшості органічних сполук на відміну від УФ-спектрів характеризуються наявністю великою кількістю ліків поглинання. Метод ІЧ-спектроскопії дає можливість одержати найбільш повну інформацію про будову і склад аналізуємої речовини, яка дозволяє ідентифікувати дуже близькі по структурі сполуки. Метод інфрачервоної спектроскопії прийнятий для ідентифікації органічних лікарських речовин з полі функціональними групами шляхом порівняння із спектрами стандартних зразків, які зняті в однакових умовах. У зв’язку з підвищеними вимогами до якості лікарських речовин ІЧ-спектроскопія, як один із найбільш надійних методів ідентифікації, мають все більше значення. Спектрофотометричне визначення проводять спектрофотометром як забарвлених, так і безбарвних сполук по вибірковому поглинання світла у видимій, ультрафіолетовій чи інфрачервоній областях спектра.
3. Задача.
Наважку вагою m = 0,300 г препарату реталоноцетату розчинили у абсолютному еталоні й отримали V( x ) = 100 мл похідного розчину, який аналізується.
Відібрали 1 мл цього розчину, додали до нього 99 мл абсолютного еталону й отримали 100 мл вимірюваного розчину. Визначити оптичну щільність А ( x ) виміряного розчину на спектрофотометрі при довжині хвилі λ = 326 нм в кюветі з товщиною поглинаючого шару Д = 1 см, яка дорівнює А ( x ) = 0,456.
Розрахувати вміст ретонолоацетату у відсотках в 1 г препарату, якщо молярний коефіцієнт гасіння спиртового розчину ретонолоацетату С23 Н32О2 рівняється: E = 50900 л* моль -1 см-1 , при 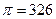 нм.
нм.
Рішення.
Відсотковий вміст х ретонолоацетату в 1 г препарату:

Де m ( x ) – вага ретонолоацетату у вихідному аналізуємому розчині яка рівняється:
m (x) = C (x) M(x) V(x),
де C (x) – концентрація ретонолоацетату у вихідному аналізуємому розчині, моль;
M (x) = 328,50 – молярна вага ретонолоацетату;
V (x) = 100 мл = 0,1 л - об’єм вихідного аналізуємого розчину.
Для визначення C (x) розрахуємо спочатку концентрацію вимірюємого розчину.
У відповідності з основним законом світопоглинання А = Ecl концентрація С = А/ El = 0,456/50,900 * 1 = 8,9 * 10 -6, моль/л. Концентрація C (x) вихідного аналізуємого розчину в 100 разів більше концентрації вимірюваного розчину:
C (x) = 100 с = 100 * 8,9 * 10 -6 = 8,9 * 10-4 , моль/л.
Тепер знаходимо M (x):
m (x) =8,9 * 10-4 * 328,50 * 0,1 = 0,0292 г.
Розрахуємо х :
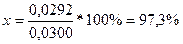 .Відповідь: 97,3 %.
.Відповідь: 97,3 %.
ЕТАЛОННА ВІДПОВІДЬ
Варіант №13
1. Якими квантовими числами описується енергетичний стан в атомі. Дати характеристику чисел.
Для електрону, який знаходиться під дією сил тяжіння до ядра, має рішення при певних значеннях енергій. Значить квантовість енергетичних станів електрону в атомі виявляється наслідком існуючих хвильових властивостей електрону та не потребує введення особливих постулатів.
Відомо, що границі атому випромінливі для електрону, тому він може знаходитись тільки всередині атому. Розглянемо модель атому, в якому електрон може здійснювати лише коливальні рухи між крайніми точками. Стан електрону в атомі характеризується деякою хвилею, відомо, що електрон в атомі стійкий. У межах атому хвильова функція Ψ (тобто амплітуда хвилі) повинна обертатись у нуль.
Квантове числа допустимі рівні енергії електрону, яке визначає значення цілого числа (n).
По моделі Бора-Резерфорда електрон у атомі мав неприливний рух з прискоренням, тобто міняв свій стан.
В одномірній моделі атому енергія електрону може приймати більш певні значення, інакше кажучи – вона квантова. Енергія електрону у атомі також величина квантова. Можливі енергетичні стани електрону в атомі визначаються величиною головного квантового числа n, яке може приймати позитивні цілочисельні значення 1, 2, 3, і т.д. Зі збільшенням n енергія електрону збільшується.
Не тільки енергія електрону в атомі може приймати певні значення. Свавільною не може бути і форма електронного облака. Вона визначається орбітальним квантовим числом l, яке може приймати цілочисельні значення від 0 до (n-1), де n – головне квантове число.
Розміри і форма електронних облаків в атомі можуть бути тільки такими, які відповідають можливим значенням квантових чисел n і 1. Орієнтація електронного облака у просторі не може бути свавільною. Вона визначається значенням третього, так званого магнітного квантового числа.
Існує ще одна величина, не пов’язана з рухом електрону навколо ядра, визначаюча його стан. Ця величина отримала назву квантового числа або S, який може стати тільки два значення і +  , у випадку останніх квантових чисел можливі значення розрізняючи на одиницю.
, у випадку останніх квантових чисел можливі значення розрізняючи на одиницю.
2. Які спектри називаються спектрами поглинання і спускання.
Спектр спускання – це спектр, утворюємий випромінюванням тіл, які світяться. Розжарені тверді та рідкі тіла дають суцільний спектр з кольорових променів, неправильно слідуючи один за одним та однаковий для всіх розжарених тіл, незалежно від хімічного складу. Розжарені гази та пари при тиску не дуже перевищуючому норму, дають лінійчатий спектр, який складається з окремих кольорових ліній розділених темними проміжками.
На відміну від розжарених твердих та рідних газів кожний хімічний елемент у стані розжарених парів має свій власний спектр.
Спектр поглинання – це зовнішній сторонній світловий потік, який пройшов через дане тіло. Для кожного елементу його лінійчатий спектр спускання та поглинання оборотні. Розташування таких ліній поглинання відповідає кольоровим лініям спускання. Всяка речовина поглинає світлове проміння тих довжин хвиль, які вона спускає у даних умовах.
3. Задача.
Розрахуйте умовний стандартний потенціал t0 хлор срібного електроду
Ag /AgCl, Cl- (електрод ІІ роду) при кімнатній температурі, якщо при тій же температурі стандартний потенціал срібного електроду Ag+ / Ag (І-го роду) дорівнює 0,799 В, а розчинність хлориду срібла Кs0(AgCl) = 1,78·10-10.
Рішення.
Потенціал хлор срібного електроду ІІ-го роду описується при кімнатній температурі рівнянням: t (Ag / AgCl, Сl ) t0 – 0,059 tg, а (Сl-).
Стандартний потенціал Е пов’язаний з постійною рівновагою:
t = 0,059 lg К, де постійна рівноваги К електрохімічної реакції:
AgCl +  H2 = Ag + Cl- + H2,
H2 = Ag + Cl- + H2,
яка полягає в ланцюзі, і який складається з хлор срібного й стандартного водородного електродів та дорівнює:
R = 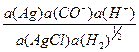 = a(Cl-)
= a(Cl-)
Активність твердих фаз:
а(Ag) = 1; а(H+) = 1;
а(Н2) » р(Н2) = 1;
Е0 = 0,059 lg а (Cl), де
а (Cl) – рівноважна активність хлор-іонів :
КS(AgCl)=a(Ag+)a(Cl-)
a(Cl-) = KS0(AgCl / aAg+).
Тоді Е0 = 0,059 lg KS0(AgCl )-0,059 lg а(Ag+ ).
Рівноважну активність катіонів срібла можна знайти, знаючи стандартний потенціал срібного спектру Ag+ / Ag..
Е = (Ag+ / Ag) = Е0(Ag+ /Ag) – 0,059 lg а(Ag+ );
Е0(Ag+ /Ag) = 0,059 lg К, де К – постійна рівновага електрохімічної реакції.
Ag+ + Н2 = Ag + Н+, яку можна знайти за формулою:
К =  =
=  ; так як
; так як
a(Ag+) = 1; a(H+) = 1; a(H2) = pH2 = 1;
Е0 = (Ag+ / Ag) = 0,059 lg ( ) = -0,059 lg a (Ag+)
Порівнюючи рівняння отримуємо
Е0 = 0,059 lg K30 (AgCl)+t(Ag / Ag) = 0,059 lg (1,78·10-10) + 0,7994 = 0,2242 В
ЕТАЛОННА ВІДПОВІДЬ
Варіант №14
1. Принцип проведення флуоресцентного аналізу.
Принцип проведення флуоресцентного аналізу базується на використанні прямої пропорційної залежності між інтенсивністю люмінесценції і концентрацією досліджу вальної речовини l = КС, де К – коефіцієнт пропорційності, але при умові, що квантовий вихід φ – const (постійна величина).
Чим більший квантовий вихід φ, тим більше коефіцієнт пропорційності і тим більше чуттєвість флуоресцентного аналізу. Тобто можна зробити висновок: флуоресцентний аналіз слід проводити в таких умовах, щоб квантовий вихід φ був максимальним і постійним.
Існують і деякі обов’язкові умови, яких треби дотримуватися і при проведенні флуоресцентного аналізу:
1. необхідно щоб виконувалися умови: довжина хвилі lавс повинна бути менше довжини хвиль люмінесценції lLm. Для збудження випромінення використовують інтервал довжин хвиль 250-800 нм.
2. аналізуємий розчин повинен бути дуже розведеним. Зменшення інтенсивності люмінесценції при збільшенні концентрації розчину називається концентраційним гасінням.
3. Усі сторонні домішки повинні бути знищеними. При їх наявності може бути посилення і флуоресцентне гасіння. До послаблення та гасіння флуоресценції веде також ї наявність розчиненого кисню.
4. температура при проведенні аналізу повинна підтримуватися постійною. При її підвищенні відбувається температура гасіння флуоресценції..
5. Коли речовина не має своєї флуоресценції, то проводить люмінесцентну реакцію – реакцію, при якій з’являється або зникає люмінесценція, або вона змінює свій колір. При проведенні цієї реакції велику роль грає природа розчину та кислотність.
2. Що таке електроди першого і другого роду.
Електроди першого роду – це електроди які обертаються за катіоном однаковим з матеріалом електродів.
Розрізняють три види електродів першого роду:
а)метал, занурений в розчин солі, також металу. На поверхні таких електродів відбувається обернена реакція: mne+ne=m.
Потенціал такого електрода першого роду залежить від активності катіонів з металу;
б)газові електроди (водородний електрод). Потенціал обертового газового водородного електроду визначається активністю іонів водорода, тобто РН розчину.
Для водородного електроду першого роду стандартний потенціал (l0 = 0) дорівнює 0, електродна реакція: Н+ + l = Н.
Число електродів, які приймають участь в реакції дорівнює: n=1;
в) амальгамні електроди уявляють собою амальгаму металу занурену в розчин, який містить катіони того ж металу. Потенціал таких спектрів залежить від активності катіонів металу в розчині реактивності металу в амальгамі. Амальгамні спектри мають високу обертаність.
Електроди другого роду обернені за іоном. Їх тип декілька видів:
а) метал, поверхня якого вкрита мало родинною сіллю того ж металу, занурений в розчин, який містить аніони, які входять у склад цієї мало родинної солі. Наприклад, хлор срібний складається зі срібної проволоки, яка вкрита мало розвинутою в воді сіллю. AgCl, занурений в водний розчин хлориду калію. На електроді відбувається обернена реакція AgCl +e = Ag + Cl-.
Електроди другого роду мають високу обертаність і стабільність в рості;
б) газові електроди другого роду.
Ці електроди в потенціометричному аналізі використовуються рідко.
Окислювально - відновлюючі електроди складаються з інертного матеріалу (золота, титану, платини), зануреного в розчин, який містить окислену і відновлену форму даної речовини. Вони бувають 2-х видів електроди, потенціал яких не залежить від активності іонів водороду, та електроди, потенціал яких залежить від активності іонів водороду.
Мембранні електроди – це електроди, обернені за тими або іншими іонами, які сорбуються твердою або рідинною мембраною. Потенціал таких електродів залежить від активності тих іонів в розчині, які сорбуються мембраною. Ці електроди мають тонку мембрану, по обидві сторони якої знаходяться різні розчини.
3. Задача.
При визначенні ванадію по методу добавок наважку сталі 0,5036 г перевели у розчин і його об’єм довели до 50,0 мл.
В дві мірні колби (50,0 мл) відібрали по 20,0 мл, в одну з них додали стандартний розчин, що містить 0,003 г ванадію, а потім в обидві колби – перекис водню. Розчини в колбах довели до мітки, профотоколориметрували і отримали Дк = 0,20 та Дк = 0,48. Розрахувати потенційний вміст ванадію.
Рішення.
Знаходимо концентрацію стандартного розчину ванадію (Сст) з урахуванням розведення:
Сст =  =
=  = 6·10-5 г/мл, де
= 6·10-5 г/мл, де
QV(ст.) – кількість ванадію в стандартному розчині, г;
V – об’єм розчину, мл..
Обчислюємо концентрацію ванадію в розчині (Сх).
Сх = Сст  г/мл
г/мл
Визначаємо кількість ванадію в наважці з урахуванням розведення розливу:
QV = 4,28·10-5  г.
г.
Процентний вміст ванадію у сталі дорівнює:
 %, де
%, де
Qcn – наважка сталі, г.
Відповідь: потенційний вміст ванадію у сталі складає 1,06 %.
ЕТАЛОННА ВІДПОВІДЬ
Варіант №15
1. Що представляє собою кондуктометричне титрування.
При кондуктометричному титруванні за ходом титрування спостерігають зміні електропровідності аналізує мого розчину, що знаходяться в кондуктометричний ніші між двома енергетичними електродами. По отриманим даним викреслюють криву кондуктометричного титрування, що відображує залежність електропровідності титрує мого розчину від об’єму титрату.
Кінцеву точку титрування частіше всього визначають екстраполяцією ділянок кривої титрування і в області зміни її нахилу. При цьому не потрібне застосування індикаторів змінюють забарвлення поблизу ТЕ.
В кондуктометричному титруванні використовують різні типи реакції: кислотно-основні; окислювально-відновні; осадові; процеси комплектування.
В залежності від того, як іони вступають в реакцію або утворюються при протіканні реакції, криві кондуктометричного титрування можуть бути різні.
По мірі титрування кислоти, електропровідність титруємого розчину дуже зменшується, так як в реакції витрачаються іони водню Н+ з високою рухомістю гідроксильних груп, що вводяться разом з титратом, також достатньо висока, але менша рухомість іонів водню.
2. Дати визначення похідної спектрофотометрії.
Похідну спектрофотометрію відносять до одного з варіантів диференціальної спектрофотометрії. Якщо в диференціальній спектрофотометрії використовують різність оптичних щільностей при одній і тій же довжині хвилі l = const (Ах = Е l (Сх – С0), то в похідній спектрофотометрії також вимірюють різність світлопоглинання, але при двох довжинах хвиль l1»l2 до ∆l - математичне похідна.
Переваги цього методу в тому, що на спектральних кривих, записаних в координатах похідна-довжина хвилі, чітко утримуються смуги, що проявляються лише в скритих max та нечітких перегинів по смузі поглинання при звичайному уявленні спектральної кривої в координатах – оптична щільність.
Вид спектральних кривих залежить від величини інтервалу ∆l, що використовується в розрахунках кривої похідної. У випадку широких смуг поглинання в похідних спектрах А=f(l) спектральну криву похідної розраховують для інтервалу ∆l = 2; 4; 6; 8 та 10 км. Оптимальний інтервал складає 4 км.
Методами похідної спектрофотометрії аналізуються також сполуки урану в присутності солей Fe, сполуки рідко земельних елементів.
3. Задача.