|
|
Химические реакции с позиций термодинамики
Подведём некоторые итоги обсуждения возможностей применения термохимии к химическим реакциям и дополнительно обратим внимание на то, что даёт термохимия и химическая термодинамика в более широком плане для понимания природы химических превращений.
Благодаря термохимии удаётся вычислить энтальпию химических реакций DН как теплоту реакций при постоянном давлении qp. В основе методики расчета лежит взаимосвязь между уравнением реакции и стандартными энтальпиями образования веществ 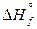 , участвующих в реакции. Определение теплоты реакций представляет собой самостоятельную задачу, т.к. некоторые из них осуществляются специально для получения теплоты.
, участвующих в реакции. Определение теплоты реакций представляет собой самостоятельную задачу, т.к. некоторые из них осуществляются специально для получения теплоты.
Однако важно знать не только тепловой эффект химических реакций. Необходимо определять, в каком направлении пойдет реакция. Интуитивно представляется вероятным, что реакция пойдет самопроизвольно в том направлении, которое приведёт к понижению энергии системы. Проявляется это изменение энергии в виде теплоты. Теплота есть форма кинетической энергии, способная передаваться от нагретого тела к холодному. Например, происходит экзотермическая реакция, температура системы повышается и теплота передается от системы в окружающее пространство. Такой процесс представляется вполне вероятным.
Действительно, многие экзотермические реакции идут самопроизвольно. Но существуют и эндотермические реакции, которые также идут самопроизвольно. Рассмотрим знакомый пример из области физических превращений:
|
Н2О(ж)  Н2О(г).
Н2О(г).
– DН°конд
DН°исп = + 44,01 кДж/моль и DН°конд= - 44,01 кДж/моль.
Хорошо известно, что при высоких температурах (лучше выше 100 °С) самопроизвольно идёт процесс испарения воды, т.е. эндотермический процесс, а при низких температурах – процесс конденсации воды, т.е. экзотермический процесс. Таким образом, оба процесса могут протекать самопроизвольно, но только при разных условиях.
Аналогическая смена направлений самопроизвольных процессов проявляется и у химических реакций. Рассмотрим реакцию
СаСО3(к) ® СаО(к) + СО2(г), DН° = + 178 кДж/моль.
Реакция – эндотермическая. Термическое разложение СаСО3(к) происходит на открытом воздухе при температурах выше 835–840 °С. Обратная реакция – экзотермическая. Она идёт самопроизвольно при низких температурах. Осадок СаСО3 образуется, например, при пропускании газообразного СО2 через раствор СаО.
Итог таков: при низких температурах более вероятны экзотермические процессы, а при высоких – возможны эндотермические процессы. Однако эти рассуждения носят качественный характер и могут привести к ошибочным выводам, когда речь идет о конкретной реакции. Энтальпия не является критерием, однозначно определяющим направление химических реакций. Кроме энтальпии введена еще одна функция состояния, названная энтропией.
Энтропия – сложное понятие,и его необходимо рассмотреть более подробно. А пока, чтобы почувствовать природу энтропии, представим себе процесс, связанный с переходом системы от упорядоченного состояния к менее упорядоченному, более вероятному. Именно за счет такого процесса энтропия системы возрастает.
То, что процесс, сопровождающийся ростом энтропии, может быть весьма вероятным, подтверждается опытным путем. Если газ находится в сжатом виде в баллоне, то его состояние является упорядоченным. Но достаточно открыть вентиль, и газ устремится наружу (за счет перепада давлений) и распределится в пределах доступного пространства. Новое состояние – менее упорядоченное, оно – более вероятное, чем исходное. Представляется невероятным, чтобы газ самопроизвольно вернулся в баллон. Следовательно, газ стремится перейти от менее вероятного упорядоченного состояния к более вероятному неупорядоченному состоянию, и этот процесс сопровождается возрастанием общего беспорядка, т.е. ростом энтропии.
Таким образом, энтропия – эта мера неупорядоченности (беспорядка) в системе.
Расширение идеального газа протекает без изменения температуры, и система не обменивается теплотой с окружающим пространством. Более того, можно избежать и работы расширения против внешнего давления, если газ поместить в одну часть системы и отделить перегородкой от другой части, в которой данного газа нет. Если разрушить перегородку, то газ заполнит все пространство системы, которая при этом не обменивается с окружающим пространством ни теплотой, ни работой. Нет обмена и веществом. Такая система является изолированной. Энтропия оказывается единственным критерием направления и равновесия процессов в изолированных системах.
Согласно второму закону термодинамики в изолированных системах возможны только такие процессы, при которых происходит рост энтропии. Когда энтропия достигает максимально возможного в данных условиях уровня, в системе наступает равновесие.
Если система не является изолированной и может взаимодействовать с окружающим пространством, то сумма изменений энтропии системы и окружения при необратимых процессах всегда положительна:
DSсистемы + DSокружения> 0.
Эта сумма равна нулю только в термодинамически обратимых процессах.
Методика расчета изменения энтропии DS° в ходе реакции аналогична той, которая используется для вычисления величины DН° реакции (см. раздел 6.7). Отличие состоит лишь в том, что в справочной литературе приводятся абсолютные значения энтропии веществ S°298, Дж /(моль×К). Тогда как абсолютные значения энтальпии не определены, абсолютные значения энтропии веществ поддаются измерению. Такую возможность дает третий закон термодинамики, который утверждает, что энтропия чистого, совершенного кристалла при 0 К равна нулю. Вблизи 0 К неупорядоченность вещества приближается к нулю. Таким путем определен уровень, относительно которого измеряют значения энтропии веществ.
В расширенном понимании третий закон термодинамики касается термодинамических свойств веществ при температуре, приближающейся к 0 К.
Возвратимся к ранее рассмотренным примерам и дадим оценку изменениям в них энтропии. При испарении воды (жидкости) образуется пар (газ), упорядоченность системы понижается, энтропия возрастает. При термическом разложении СаСО3(к) образуется СО2(г) и энтропия возрастает. Очевидно, обратные процессы должны сопровождаться уменьшением энтропии. Но они также протекают самопроизвольно в определенных условиях. Следовательно, ни энтальпия и ни энтропия не могут порознь предсказать направление таких процессов.
Определим, к какому типу систем относятся те, в которых происходят эти процессы. Рассматриваемые процессы сопровождаются тепловыми эффектами, а системы, в которых они протекают, могут обмениваться с окружающим пространством, по крайней мере, теплотой. Следовательно, системы не являются изолированными, их можно отнести к типу закрытых систем. Для определения направления и равновесия процессов в закрытых системах требуются дополнительные функции состояния, такие как энергия Гельмгольца и энергия Гиббса.
Энергия Гельмгольца характеризует процессы в системах при постоянных Т и V (изохорно-изотермические процессы). Её определяют по уравнению
D А° = DU° – ТD S°, (6.12)
где D А° – энергия Гельмгольца реакции при стандартных условиях; DU° – изменение внутренней энергии системы в результате реакции при стандартных условиях; DS° – изменение энтропии в результате реакции при стандартных условиях; Т – температура, при которой происходит реакция, К.
Уравнение (6.12) является важным для теории и практики.
Энергия Гиббса относится к процессам в системах при постоянных Т и Р (изобарно-изотермические процессы) и её определение следует из уравнения
DG° = DH° – TDS°, (6.13)
где DG°– энергия Гиббса реакции при стандартных условиях; DH° – изменение энтальпии в результате реакции при стандартных условиях. Величины D А°, DU°, DH° и DS° принято определять при 298 К.
На практике чаще встречаются изобарно-изотермические процессы и энергия Гиббса используется чаще, чем энергия Гельмгольца. Энергию Гиббса называют также изобарно-изотермическим потенциалом и свободной энергией.
Энергия Гиббса реакции DG – функция состояния системы, экстенсивный параметр. Все параметры, входящие в уравнение (6.13), относятся непосредственно к системе. Энергия Гиббса является критерием, определяющим вероятность и направление химических реакций в системах, которые не обмениваются с окружающей средой массой, но могут обмениваться энергией (в форме теплоты и работы). Самопроизвольные процессы сопровождаются понижением энергии Гиббса. Если в ходе химической реакции энергия Гиббса достигает минимально возможного в данных условиях уровня, то наступает химическое равновесие.
Обобщая сказанное, можно сформулировать следующие выводы:
если DG < 0, то возможен самопроизвольный процесс;
если DG = 0, то наступает состояние равновесия;
если DG > 0, то процесс в прямом направлении невозможен, однако для обратного процесса будет выполняться соотношение DG < 0.
Величина DG характеризует способность системы совершать полезную работу (без расширения). Поскольку работа, совершаемая системой, максимальна в обратимом процессе, то –DG = –wобр. Отсюда DG – максимальная величина работы системы против окружения при постоянных Т и Р. Пример – электрическая работа.
Энергия Гиббса реакции DG позволяет не только определять направление химических реакций, но и оценивать полноту их прохождения, что основывается на взаимосвязи между энергией Гиббса и константой равновесия реакции K. На примере идеальных газов показано, что
DG = DG° + RT ln Q, (6.14)
где Q – отношение парциальных давлений газов (раздел 8.3); DG° – энергия Гиббса реакции при стандартных условиях.
Так, для реакции
N2(г) + 3H2(г) ®2NH3(г)
выражение для Q имеет следующий вид:
 (6.15)
(6.15)
В условиях равновесия DG = 0, и уравнение (6.14) принимает вид
 (6.16)
(6.16)
Но отношение равновесных парциальных давлений является выражением константы равновесия (раздел 7.4).
 (6.17)
(6.17)
Отсюда
DG° = –RTlnKР (6.18)
Это же уравнение запишем в форме антилогарифма:
 (6.19)
(6.19)
Комбинируя уравнения (6.14) и (6.18), можно получить
 (6.20)
(6.20)
или
 (6.21)
(6.21)
Таким образом, отношение Q / KР показывает, как далека реакция от состояния равновесия. Для этого нужно знать парциальные давления газов в исходном состоянии и вычислить значение Q. Если Q < KР, то согласно уравнению (6.21) DG < 0 и реакция будет протекать в прямом направлении, т.е. в сторону понижения энергии Гиббса. В условиях равновесия величина КР характеризует отношение равновесных значений парциальных давлений газов. Сравнивая исходное и равновесное состояния, можно вычислить выход реакции.
Анализ уравнения (6.20) позволяет также сделать заключение, что при равенстве абсолютных значений  достигается DG = 0 и в системе устанавливается состояние химического равновесия. Это свидетельствует о равенстве энергий Гиббса реагентов и продуктов реакции. Другой вывод состоит в том, что при стандартных условиях, когда парциальные давления газов или концентрации веществ равны 1 атм или 1 М, соответственно
достигается DG = 0 и в системе устанавливается состояние химического равновесия. Это свидетельствует о равенстве энергий Гиббса реагентов и продуктов реакции. Другой вывод состоит в том, что при стандартных условиях, когда парциальные давления газов или концентрации веществ равны 1 атм или 1 М, соответственно  и DG = DG°. Такой результат вполне закономерен, когда в исходном состоянии система оказывается в стандартном состоянии.
и DG = DG°. Такой результат вполне закономерен, когда в исходном состоянии система оказывается в стандартном состоянии.
Вычисление величины DG° реакции можно проводить по уравнению
 (6.22)
(6.22)
если известны стандартные энергии Гиббса  образования каждого вещества – участника реакции. Значения
образования каждого вещества – участника реакции. Значения  для многих веществ приводятся в справочной литературе. Отметим, что энергия Гиббса – экстенсивный параметр и в расчетах необходимо учитывать число молей вещества, вступающего в реакцию. Если для расчета берут молярные значения энергии Гиббса образования веществ, то вместо числа молей принимают стехиометрические коэффициенты перед веществами в уравнении реакции. Энергии Гиббса простых веществ в стандартном состоянии приняты равными нулю.
для многих веществ приводятся в справочной литературе. Отметим, что энергия Гиббса – экстенсивный параметр и в расчетах необходимо учитывать число молей вещества, вступающего в реакцию. Если для расчета берут молярные значения энергии Гиббса образования веществ, то вместо числа молей принимают стехиометрические коэффициенты перед веществами в уравнении реакции. Энергии Гиббса простых веществ в стандартном состоянии приняты равными нулю.
При вычислении величины DG° можно вначале определить значения DН° и DS° реакции, а затем их подставить в уравнение (6.13) и вычислить величину DG°.
Если температура Т, при которой протекает реакция, не очень сильно отличается от 298 К, то величину  вычисляют по уравнению (6.13), подставляя в него величины DН
вычисляют по уравнению (6.13), подставляя в него величины DН  и DS и фактическое значение Т. Но при необходимости проведения точных расчетов учитывают зависимости функций состояния от температуры. При близких значениях Т1 и Т2, когда Т1 < Т2, зависимостями DН и DS от Т можно пренебречь, и решение системы двух уравнений:
и DS и фактическое значение Т. Но при необходимости проведения точных расчетов учитывают зависимости функций состояния от температуры. При близких значениях Т1 и Т2, когда Т1 < Т2, зависимостями DН и DS от Т можно пренебречь, и решение системы двух уравнений:

приведет в выражению

где K1 при Т1 и K2 при Т2.
После преобразований этого выражения получаем уравнение
 (6.23)
(6.23)
Уравнение (6.23) позволяет вычислить значение K2 при заданном значении Т2, если известно значение K1 при Т1. Из этого уравнения следует также, что влияние температуры на величину константы равновесия зависит как от знака величины DН реакции, так и от ее абсолютного значения. Например, для экзотермической реакции (DН < 0) при повышении температуры (Т2 > Т1) установится соотношение  соответственно уменьшится величина константы равновесия (K2 < K1) и, следовательно, понизится выход реакции.
соответственно уменьшится величина константы равновесия (K2 < K1) и, следовательно, понизится выход реакции.
Пример 6.10. Дана химическая реакция
СаСО3(к) = СаО(к) + СО2(г).
Определим направление этой реакции при температурах 700 и 1000 °С. Если направление реакции изменяется с повышением температуры, то дадим оценку температуры, при которой происходит смена направления реакции. Зависимость термодинамических функций от температуры
учитывать не будем.
Решение. Из справочной литературы выпишем значения  и
и  веществ – участников процесса:
веществ – участников процесса:
| ,кДж / моль
|  , Дж/(моль×К) , Дж/(моль×К)
| |
| СаСО3(к) | –1207 | |
| СаО (к) | –635 | |
| СО2(г) | –393,5 |
Вычисления проведем по той методике, которая применена в примере 6.9.
При температуре 700 °С (973 К )


Несмотря на существенное отклонение температуры процесса от стандартной, для оценочного расчета величины воспользуемся стандартными значениями  и
и  .
.
= – Т  =
=

Для обратной реакции = – 22  . Следовательно, при Т = 700 °С (973 К) реакция идет в обратном направлении.
. Следовательно, при Т = 700 °С (973 К) реакция идет в обратном направлении.
Воспользуемся вычисленными значениями и и проведем вычисление величины при температуре 1000 °С (1273 К ).
= – Т =

При Т = 1273 К реакция идет в прямом направлении. Смена направления реакции при повышении температуры подтверждается.
Дадим оценку температуры, при которой происходит изменение направления реакции. Очевидно, при такой температуре реакция в стандартных условиях (Р = 1 атм) меняет направление, когда K = 1 и = 0. Отсюда
= – Т = 0
или

Эта температура близка к той, выше которой действительно начинается термическое разложение СаСО3(к).
Пример 6.11. Для реакции
2NO(г) + O2(г) = 2NO2(г)
вычислим значения DН°, DS°, DG° и KP (при 298 К ) и определим направление реакции, если в исходном состоянии PNO = 1,2 атм, РО2= 0,5 атм и РNO2= 10,0 атм.
Решение. Выпишем из справочной литературы значения и  для веществ, участвующих в реакции:
для веществ, участвующих в реакции:
| NO(г) | O2(г) | NO2(г) | |
| , кДж / моль
| |||
| , Дж / (К×моль)
|


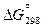 =
=  – Т
– Т  =
=
= –112 кДж – 298К×(–147 Дж / К)×1 кДж / 1000 Дж = – 68 кДж.
Вычисление величины Kр проведем по уравнению (5.19):

Вычислим величину Qр для заданных парциальных давлений газов:
 .
.
Поскольку Qр << Kр, то реакция находится далеко от состояния равновесия и должна протекать в прямом направлении.
Комментарий: при вычислении величины KP использовано значение R = 0,00831 кДж /К, в то время как в справочной литературе приводится значение R = 8,31 Дж/(моль×К). Как видно, нами учитывался фактор пересчета 1 кДж/1000 Дж и принималось во внимание, что все использованные величины до вычислений были отнесены к 1 моль (молярные величины).
Возникает также вопрос об единицах измерения Q. Величины Q и KP обычно берутся как безразмерные.
Пример 6.12. Рассмотрим следующее равновесие
PbO + CO Pb + CO2; DH  = – 63,7 кДж/моль. При Т1 = 298 К константа равновесия KP = 49,7. Вычислим значение KP для этой реакции при Т2 = 398 К.
= – 63,7 кДж/моль. При Т1 = 298 К константа равновесия KP = 49,7. Вычислим значение KP для этой реакции при Т2 = 398 К.
Решение. Вычисления проведем на основе уравнения (6.23):
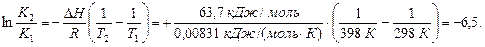 K2 = 7,4 · 10-2.
K2 = 7,4 · 10-2.
Ответ: K398 = 7,4×10–2. Реакция является экзотермической. С повышением температуры величина константы равновесия уменьшается и выход реакции понижается.
___________________________________________________________________
Качественная оценка влияния температуры на направление химических реакций может быть выполнена при использовании стандартных значений DН° и . Пренебрегая зависимостью этих функций от температуры, температурный вклад учитывают за счет произведения Т  . При этом знак и величина
. При этом знак и величина  будут определяться как величинами, так и знаками DН° и Т×
будут определяться как величинами, так и знаками DН° и Т×  . Для конкретных реакций при заданных значениях Т проводят расчеты величин
. Для конкретных реакций при заданных значениях Т проводят расчеты величин  , которые позволяют делать оценочные выводы о направлении реакций.
, которые позволяют делать оценочные выводы о направлении реакций.
В качестве примеров выбрано несколько химических реакций. Для них вычислены стандартные значения DН°, DS°, и DG° (Т = 298 К), которые обобщены в табл. 6.3.
Итак, обратим внимание на знаки DН° и DS°. Возможны четыре варианта их сочетания, что отражено в реакциях 1–4. Типичными для химических реакций являются первые два варианта из указанных в табл. 6.3. Допустим (первый вариант), что величины DН° и DS° имеют одинаковый отрицательный знак. Тогда в уравнении энергии Гиббса DН° < 0 и –T×DS° > 0. Как известно, реакция идет слева направо при DG°< 0, т.к. K > 1. В наших условиях «энтальпийный фактор» способствует прямой реакции, а «энтропийный фактор» помогает реакции идти в обратном направлении. Суммарное влияние обоих факторов таково, что при низких температурах основной вклад в изменение свободной энергии будет вносить «энтальпийный фактор» и реакция приобретет прямое направление. Но при повышении температуры произведение ТDS возрастает и наступает момент, когда |TDS°| > |DН°|. Превалирующим становится «энтропийный фактор», и с учетом отрицательного знака DS° изменится знак величины DG°, реакция преодолеет состояние равновесия и пойдет в обратном направлении. Примером может служить первая реакция из приведенных в табл. 6.3.
Рассмотрим второй вариант: знаки DН° и DS° опять одинаковые, но положительные. Оценку знака величины DG° проведем так же, как в первом варианте, но вывод получим противоположный. При низких температурах реакция пойдет в обратном направлении (DН° > 0), а при высоких – в прямом (–ТDS° < 0). Такой вариант часто реализуется при разложении сложных веществ на простые, особенно если разложение сопровождается выделением газов. Энтропия системы должна возрастать, что способствует самопроизвольному течению реакции (когда «энтальпийный фактор» перестает компенсировать рост произведения ТDS°). Пример – вторая реакция в табл. 6.3.
Т а б л и ц а 6.3. Набор химических реакций, характеризующихся
разными знаками величин DН° и DS°
| Номер реакции | Химическая реакция | DН°, кДж | DS°, Дж/К | DG°, кДж |
| 2SO2(г) + O2(г) ® 2SO3(г) | –198 | –188 | –142 | |
| CaCО3(к) ® CaO(к) + CO2(г) | +178 | +159 | + 131 | |
| С6Н12О6(к) ® 2С2Н5ОН(ж) + 2СО2(г) | – 67 | +538 | –227 | |
| 3О2(г) ® 2О3(г) | +286 | –137 | +326 | |
| СН4(г) + 2О2(г) ® СО2(г) + 2Н2О(г) | –803 | – 4 | –802 |
Следующие два варианта встречаются значительно реже. Третья реакция должна идти в прямом направлении независимо от температуры, т.к. DН° < 0 и –ТDS° < 0. Четвертая реакция не идет самопроизвольно ни при каких температурах, т.к. DН° > 0 и –ТDS° > 0. Как известно, озон образуется при электрическом разряде через слой кислорода. Таким путем достигается повышение свободной энергии системы, что не происходит самопроизвольно.
Встречаются также варианты, когда в ходе реакции энтропия системы почти не изменяется. Это может происходить, например, когда число молей газов в продуктах реакции и в исходных веществах одинаково. Примером может служить пятая реакция в табл. 6.3. Очевидно, влияние температуры на направление таких реакций становится минимальным.
* В так называемой термохимической системезнаков тепловой эффект экзотермической реакции считают положительным, а эндотермической реакции – отрицательным. В настоящее время термохимическая система знаков не рекомендована к употреблению.