|
|
Проявления мезомерного эффекта
Полярность молекулы.Смешение π-электронной плотности к более электроотрицательному атому (мезомерный эффект) происходит гораздо эффективнее и распространяется на значительно большее расстояние, чем смещение σ-электронной плотности (индуктивный эффект). В результате молекула поляризуется сильнее. Это видно, например, при сравнении дипольных моментов бутаналя и его ненасыщенного аналога - кротонового альдегида:

Стабильность катионов.Аллильный карбокатион необычайно стабилен: на 280 кДж/моль устойчивее метильного карбокатиона и примерно так же стабилен, как третичный бугильный катион.

Причина этого явления - делокализация положительного заряда благодаря сопряжению вакантной (пустой) p-орбитали карбокатионного центра с соседней двойной связью. Связь С=С здесь выступает как донор π-электронов, т.е. группа, проявляющая +М-эффект по отношению к акцептору - sp2-гибридизованному плоскому карбокатионному центру. Необходимое условие сопряжения -параллельность р-орбиталей - приводит к тому, что все заместители (на приведенном рисунке - атомы водорода) лежат в той же плоскости, что и три атома углерода. Эти три атома С связаны "частично двойными" связями. Центральный атом углерода не имеет заряда, а оба крайних формально несут по половине положительного заряда. Такое распределение часто приводит к аллильной перегруппировке, суть которой состоит во взаимодействии нуклеофила с любым из крайних атомов промежуточно образующегося в реакции аллил-катиона.
В качестве примера рассмотрим присоединение НСl к 2,4-гексадиену, приводящее к 4-хлор-2-гексену и 2-хлор-3-гексену. Образование этих продуктов показывает, что водород присоединяется к С-2 с образованием карбокатиона А, а не к С-3 с образованием карбокатиона В:

Оба карбокатиона - вторичные, но А еще и аллильный, а следовательно, значительно более устойчивый. Нуклеофильная атака хлорид-аниона по крайним атомам аллильной системы приводит к двум изомерным продуктам.
Еще большая стабилизация карбокатиона за счет сопряжения наблюдается при наличии соседнего ароматического кольца. Для бензильного катиона можно  построить уже не две, а четыре предельные резонансные структуры, три из которых близки по энгергии. Согласно принципам теории резонанса это соответствует значительной стабильности системы. В кольце заряд распределяется преимущественно на орто- и пара-положения.
построить уже не две, а четыре предельные резонансные структуры, три из которых близки по энгергии. Согласно принципам теории резонанса это соответствует значительной стабильности системы. В кольце заряд распределяется преимущественно на орто- и пара-положения.
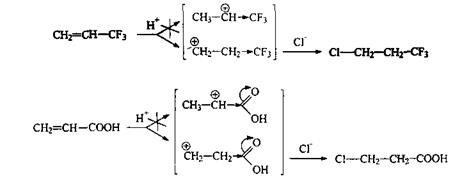 Если при двойной связи алкена есть сильные электроноакцепторные группы (-I, -М-эффект), то присоединение галогенводородов к ним происходит против правила Марковникова:
Если при двойной связи алкена есть сильные электроноакцепторные группы (-I, -М-эффект), то присоединение галогенводородов к ним происходит против правила Марковникова:
Электроноакцепторный заместитель дестабилизирует расположенный рядом катион (концентрация заряда), поэтому карбокатионный центр стремится образоваться от него подальше. Однако, гидрогалогенирование винилгалогенидов (–I-заместитель !) подчиняется правилу Марковникова. Причина - стабилизация рядом расположенного катиона сопряжением его с неподеленными электронными парами галогена (противоположно направленный +М-эффект):

Кислотность. Стабильность анионов.Важнейшим фактором, определяющим протонную кислотность, является стабильность аниона, образующегося при отщеплении протона. Значительно более высокая степень диссоциации карбоновых кислот
 (рКа ≈ 4,8) по сравнению со спиртами(рКа ≈ 16) обусловлена тем, что анион кислоты, в отличие от алкоголят-аниона, стабилизирован благодаря делокализации отрицательного заряда посредством -I- и -М-эффектов полярной связи С=O. Второй эффект особенно важен.
(рКа ≈ 4,8) по сравнению со спиртами(рКа ≈ 16) обусловлена тем, что анион кислоты, в отличие от алкоголят-аниона, стабилизирован благодаря делокализации отрицательного заряда посредством -I- и -М-эффектов полярной связи С=O. Второй эффект особенно важен.
При этом отрицательный заряд равномерно распределен между обоими атомами кислорода (примерно по 1/2е), но отсутствует на среднем атоме углерода. –I и –М-эффекты заместителя R также способствуют делокализации отрицательного заряда (частичному смещению его на R), что повышает стабильность аниона.
Аналогично объясняется повышенная сравнительно с алифатическими спиртами кислотность фенолов(PhOH pKa ≈ 9,95): ароматическое кольцо принимает на себя часть отрицательного заряда, который распределяется между атомами цикла, причем преимущественно - в орто- и пара-положения:

Подчеркнем двойственное поведение фенильной группы: здесь, в сопряжении с сильным донором электронов (–О–) она проявляет отрицательный мезомерный эффект (-М), а в бензильном катионе, в сопряжении с сильным акцептором (С+), - положительный (+М).
Введение электроноакцепторных заместителей, особенно обладающих -М-эффектом, в орто- и пара-положения ароматического кольца резко усиливает делокализанию заряда, и следовательно, кислотность фенола [для п-(O2N)C6H5OH pKa = 7,1]:

Электроноакцепторные заместители усиливают кислотность также бензойных кислот, но их влияние осуществляется посредством -I-эффекта, а -М-эффект в полную силу не действует, так как цепь сопряжения не достигает отрицательно заряженного атома кислорода.

Исключительно важную роль играют отрицательные мезомерный и индуктивный эффекты в стабилизации карбанионов. Проявляется это в степени диссоциации связи С–Н (термодинамическая кислотность) и скорости ее ионизации (кинетическая кислотность). В общем случае, чем стабильнее образующийся карбанион, тем сильнее и быстрее ионизуется связь С–Н.
 Алканы не способны к отщеплению протона (величина рКа для них определяется с помощью специальных экстраполяции). Заместители с большим -М-эффектом, присоединенные к тому же углеродному атому, делают диссоциацию С–Н С– + Н+ возможной, поскольку стабилизируют карбанион, эффективно делокализуя заряд:
Алканы не способны к отщеплению протона (величина рКа для них определяется с помощью специальных экстраполяции). Заместители с большим -М-эффектом, присоединенные к тому же углеродному атому, делают диссоциацию С–Н С– + Н+ возможной, поскольку стабилизируют карбанион, эффективно делокализуя заряд:

| Соединение | рКа | Соединение | рКа |
| СН4 | СН3(СН2)nСН3 | >40 | |
| CH3NO2 | 10,2 | СН3С(=O)СН3 | |
| CH2(NO2)2 | 4.0 | СН2(СО2Еt)2 | 13,3 |
| CH(NO2)3 | ≈ 0 | CH3C(=O)CH2 COOC2H5 | 10,7 |
| CH3C(=O)CH2C(=O)CH3 | 8,8 |
При увеличении числа таких заместителей делокализация заряда усиливается и кислотность растет (см. таблицу)
Галогены, благодаря –I-эффекту, также должны стабилизировать карбанион. Это подтверждается сравнением скоростей образования карбанионов ацетона и его хлор- и дихлорпроизводных (см. следующую таблицу).
| Соединение | Относит, скорость образования карбанионов |
| СН3С(=O)СН3 | |
| CH3C(=O)CH2Cl | 1,2·102 |
| СH3C(=O)CHCl2 | 1,6 ·103 |
| CH3(=O)CH2COOC2H5 | 2,6·106 |
| СН3С(=O)СН2С(=O)СН3 | 3,6 ·107 |
Особенно легко образуются карбанионы, стабилизированные мезомерным эффектом двух карбонильных групп. Например, 2,4-пентандион (ацегилацетон) образует анион в 3,6 107 раз быстрее, чем ацетон:
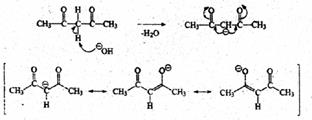
Из приведенной схемы видно, как в анионе 2.4-пентандиона обе карбонильные группы участвуют в делокализации отрицательного заряда. Отметим, что отрицательный заряд в анионе не равномерно распределен между атомами С и О: конечно, он более сосредоточен на электроотрицательном кислороде. Поэтому при обсуждении реального строения таких частиц правильнее говорить не о карбанионах, а о енолят-анионах.
Основность, то есть способность присоединять протон (в теории Бренстеда), увеличивается с увеличением плотности отрицательного заряда на атоме. Поэтому сила оснований тем больше, чем сильнее электронодонорные свойства заместителей у центрального атома, и тем меньше, чем сильнее их электроноакцепторные свойства.
Описанная выше стабилизация анионов А– при делокализации отрицательного заряда благодаря -М-эффекту заместителей объясняет повышение кислотности их сопряженных кислот НА (не путать с сопряжением π-электронов !). Одновременно это означает понижение основности анионов А–.
Увеличение основности аминов благодаря +I-эффекту заместителей и уменьшение ее из-за –I-эффекта описано на стр.12-13. Еще большее воздействие оказывают заместители с
–М-эффектом, поскольку они делокализуют неподеленную электронную пару азота, вовлекая ее в сопряжение с полярными π-связями (n-π-сопряжение). Рассмотрим это явление на нескольких примерах, используя для количественной оценки эффекта величины рКа сопряженныx кислот R3NH+ (чем больше рКа, тем больше основность R3N).
Резкое уменьшение основности наблюдается, если аминогруппа
находится рядом с карбонильной группой (-М, - I-эффекты):

В то время, как этиламин – довольно сильное основание, ацетамид основными свойствами практически не обладает.
Сравнение основности циклогексиламина, анилина и аммиака (рКа 9,25) показывает, что алкильные заместители при атоме азота
 увеличивают силу основания (+I-эффект), а арильные уменьшают (-М- и -I-эффекты).
увеличивают силу основания (+I-эффект), а арильные уменьшают (-М- и -I-эффекты).
Протонирование азота в анилине приводит к катиону PhNH3+, в котором отсутствует неподеленная пара электронов, т.е. отсутствует и n-π-сопряжение, что энергетически невыгодно.
Приведенные для анилина резонансные структуры показывают, что электроотрицательные заместители, особенно в орто- и пара-положениях, должны сильно снижать основность ариламинов.

Данные для нитроанилинов подтверждают это. Особенно мала основность о-производного, что объясняется заметным влиянием -I-эффекта и дополнительной стабилизацией структуры с разделенными зарядами за счет внутримолекулярной водородной связи.
Если необходимое условие для действия М-эффекта (для сопряжения) - параллельность р- и n-орбиталей - невыполнимо, то этот эффект не проявляется. Так, N,N-диметил-2,4,6-тринитроанилин в 40000 раз основнее, чем 2,4,6-тринитроаннлин (рКа больше на 4,6). Это огромное различие нельзя объяснить только +I-эффектом
 метильных групп. Например, для про тонированных форм анилина (рКа = 4,6) и N,N-диметиланилина (рКа = 5,2) различие невелико.
метильных групп. Например, для про тонированных форм анилина (рКа = 4,6) и N,N-диметиланилина (рКа = 5,2) различие невелико.
Основная причина заключается в том, что объемистые метильные группы не могут разместиться в одной плоскости с двумя соседними нитрогруппами. Группа N(CH3)2 вынуждена повернуться относительно плоскости ароматического кольца. При этом орбиталь неподелен-ной пары на тгоме азота не может быть перпендикулярной этой плоскости, т.е параллельной р-орбиталям кольца. n-π-Сопряжение нарушается, и -М-эффект тринитрофенильного заместителя "выключается". Снижение основности происходит уже только под влиянием -I-эффекта нитрогрупп, передающегося через σ-связи кольца.
 Электрофильное замещение в ароматическом ряду.Ключевой стадией, которая определяет скорость реакции, является образование σ-комплекса. Эта положительно заряженная частица представляет собой наиболее высокий по энергии интермедиат данного процесса, что связано с нарушением ароматичности при присоединении электрофила Е+. Электронодонорные заместители (+I, +М) должны стабилизировать с-комплекс, т.е. понижать его энергию, что означает ускорение реакции. Электроноакцепторные -наоборот. Поскольку заряд в σ-комплексе распределен, в основном, на орто- и пара-положения относительно вновь вступающего заместителя Е, то именно здесь донорные группы окажут максимальное стабилизирующее воздействие, а акцепторные - максимально дестабилизирующее.
Электрофильное замещение в ароматическом ряду.Ключевой стадией, которая определяет скорость реакции, является образование σ-комплекса. Эта положительно заряженная частица представляет собой наиболее высокий по энергии интермедиат данного процесса, что связано с нарушением ароматичности при присоединении электрофила Е+. Электронодонорные заместители (+I, +М) должны стабилизировать с-комплекс, т.е. понижать его энергию, что означает ускорение реакции. Электроноакцепторные -наоборот. Поскольку заряд в σ-комплексе распределен, в основном, на орто- и пара-положения относительно вновь вступающего заместителя Е, то именно здесь донорные группы окажут максимальное стабилизирующее воздействие, а акцепторные - максимально дестабилизирующее.
Дестабилизация электроноакцепторным заместителем будет минимальной, если атака электрофила Е направлена на мета-положение кольца.


В результате, ароматические соединения, имеющие электронодонорный заместитель, реагируют легче бензола и дают преимущественно орто- и пара-дизамещенные продукты
(заместители I рода). Те, что содержат электроноакцепторный
заместитель, реагируют труднее бензола и дают мета-дизамещенные производные (заместители II рода).
Особый случай составляют галогены. Как индуктивные акцепторы они дезактивируют ароматическое ядро к электрофильному замещению, но как мезомерные доноры –стабилизируют карбокатионный центр на ближайшем атоме углерода, т.е. ориентируют электрофил и орто- и пара-положение.