|
|
Особливості морфологічної діяльності мозку.
♦ Мікрогірія
 - аномалія розвитку головного мозку, що характеризується малими розмірами мозкових звивин при збільшеному їх числі. Пояснюється зупинкою розвитку мозку на 6-7 місяці внутрішньоутробного життя. Найчастіше зустрічається при мікроцефалії, поренцефалії. Даний дефект розвитку мозку супроводжується вираженою затримкою фізичного і психічного розвитку людини.
- аномалія розвитку головного мозку, що характеризується малими розмірами мозкових звивин при збільшеному їх числі. Пояснюється зупинкою розвитку мозку на 6-7 місяці внутрішньоутробного життя. Найчастіше зустрічається при мікроцефалії, поренцефалії. Даний дефект розвитку мозку супроводжується вираженою затримкою фізичного і психічного розвитку людини.
♦ Пахігірія -розширення звивин великих півкуль мозку і зменшення кількості мозкових борозен. У деяких відділах мозку борозни відсутні і звивини взагалі не розрізняються (Агірія). Вага головного мозку, як правило, зменшена. Клітини рухової кори часто залишаються на стадії ембріонального розвитку. Клінічно визначається затримка загального розвитку, повільний, хореоатенозний характер спонтанних рухів, пригнічення рефлекторної діяльності.
♦ Мікроцефалія
 - значне зменшення розмірів черепа і відповідно головного мозку при нормальних розмірах інших частин тіла. Мікроцефалія супроводжується розумовою недостатністю - від нерізко вираженої імбецильності до ідіотії.
- значне зменшення розмірів черепа і відповідно головного мозку при нормальних розмірах інших частин тіла. Мікроцефалія супроводжується розумовою недостатністю - від нерізко вираженої імбецильності до ідіотії.
Причинами микроцефалии можуть бути різні фактори: радіація, інфекції, ліки, генетичні порушення та ін Причини вродженої мікроцефалії - внутрішньоутробні інфекції такі, як краснуха, цитомегаловірус, токсоплазмоз. Мікроцефалія характерна для таких синдромів, як:
-трисомія по 18 хромосомі (синдром Едвардса);
-трисомія по 13 хромосомі (синдром Патау);
-синдром котячого крику;
-синдром Міллера;
-синдром Прадера-Віллі та ін;
-фетальний алкогольний синдром.
Як такого, спеціального лікування мікроцефалії не існує. Лікування спрямоване на зняття виникаючих симптомів, т.е є симптоматичним.
♦ Макроцефалія
 -пропорційне збільшення всього мозку без головного водянки, яке може протікати безсимптомно, але завжди з відставанням розумового розвитку. Зовнішній вигляд мозку зазвичай мало відрізняється від нормального. Дослідження спиномозговой рідини не виявляє патологічних змін, тиск нормальний. Форма голови зазвичай нормальна, без опуклостей і нависання лобових кісток, характерних для гідроцефалії. Велике джерельце закривається з запізненням, часто він широко відкритий, але не вибухає. Макроцефалія може бути вродженою, але може також виникнути і після народження, причини невідомі.
-пропорційне збільшення всього мозку без головного водянки, яке може протікати безсимптомно, але завжди з відставанням розумового розвитку. Зовнішній вигляд мозку зазвичай мало відрізняється від нормального. Дослідження спиномозговой рідини не виявляє патологічних змін, тиск нормальний. Форма голови зазвичай нормальна, без опуклостей і нависання лобових кісток, характерних для гідроцефалії. Велике джерельце закривається з запізненням, часто він широко відкритий, але не вибухає. Макроцефалія може бути вродженою, але може також виникнути і після народження, причини невідомі.
♦ Гідроцефалія
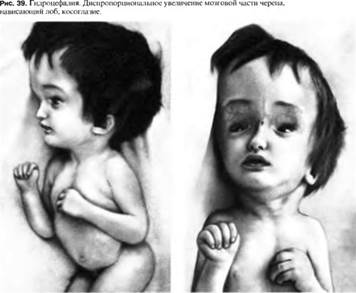 - захворювання, що характеризується надлишковим накопиченням цереброспінальної рідини у шлуночковій системі головного мозку в результаті ускладнення її переміщення від місця секреції (шлуночки головного мозку) до місця абсорбції до кровоносної системи (субарахноїдальний простір) — окклюзійна гідроцефалія, або в результаті порушення абсорбції — арезорбти́вна гідроцефалія.
- захворювання, що характеризується надлишковим накопиченням цереброспінальної рідини у шлуночковій системі головного мозку в результаті ускладнення її переміщення від місця секреції (шлуночки головного мозку) до місця абсорбції до кровоносної системи (субарахноїдальний простір) — окклюзійна гідроцефалія, або в результаті порушення абсорбції — арезорбти́вна гідроцефалія.
У більшості випадків, виникнення гідроцефалії у новонародженого зумовлено інфекційним захворюванням, що перенесла мати під час вагітності (цитомегаловірусна інфекція), що призводить до порушення роботи шлуночкової системи головного мозку плоду. Це призводить до ускладнення циркуляції ліквору та/або його надлишкової продукції. Окрім вродженої гідроцефалії, може розвинутись (частіше всього у перші місяці життя новонародженого) й набута гідроцефалія після перенесених менінгітів, менінгоенцефалітів, травм голови, інтоксикацій тощо. Порушення циркуляції цереброспінальної рідини веде до підвищення внутрішньочерепного тиску й так званого гіпертензійно-гідроцефального синдрому. В результаті тиску на ділянки мозку починає падати зір, виникають судоми, стискання стовбуру головного мозку проявляється розладами руху очей (косоокість, парез погляду вгору (симптом «сонця, що заходить»)), слабкістю у верхніх та нижніх кінцівках. Це може призвести до смерті, важких неврологічних розладів, зниження інтелектуальних здібностей.
♦ Гомоцистеїнурія- нестача цистатіонін-б синтази впливає на перетворення метіоніну (Met) в цистеїн (Cys).Накопичення гомоцистеїну, метіоніну.Уражені діти безсумнівно є нормальними. Клініка виникає через 1-2 роки, коли виявляються незворотньо ураженими різні системи: ектопія кришталика, глаукома, тромбоемболія, остеопорозу, розумове відставання, параліч.
Лікування: спеціальна дієта без метіоніну; піридоксин.
♦ Галактозимія -рідкісне генетичне порушення обміну речовин, яке порушує нормальний процес метаболізму вуглеводу (цукру) галактози. Успадковується за аутосомно-рецесивним типом і виникає через дефіцит активності ферменту, який відповідає за засвоєння організмом галактози.
ПРИЧИНИ ВИНИКНЕННЯ:
При нормальному метаболізмі, лактоза, яка міститься в продуктах харчування (наприклад, у молочних продуктах) під дією ферменту лактази, розщеплюється, утворюючи глюкозу і галактозу. У осіб, хворих на галактоземію, ферменти, необхідні для перетворення галактози або відсутні, або ж їх рівень дуже низький, що призводить до накопичення токсичного галактозо-1-фосфату в різних тканинах (як і у випадку класичної галактоземії), ці процеси призводять до гепатомегалії (збільшення печінки), цирозу печінки, ниркової недостатності, катаракти, пошкодження головного мозку і яєчників. Без лікування, смертність дітей грудного віку із діагнозом галактоземія становить близько 75%.
Галактоземія успадковується за аутосомно-рецесивним типом, тобто дитина буде хворої тільки у тому випадку, якщо успадкує дві копії дефектного гена (по одній від кожного з батьків). Гетерозиготні особи вважаються носіями хвороби, адже вони успадковують один нормальний ген і один дефектний ген. Але, у носіїв, як правило, теж проявляються деякі симптоми галактоземії, проте, звісно, у дуже помірній формі.
Довгострокові наслідки галактоземії:
- порушення мови;
- атаксія – розлад координації довільних рухів;
- дисметрія;
- зменшення щільності кісток;
- передчасна менопауза;
- катаракта.
♦ Фруктозурія - спадкове захворювання, обумовлене недоліком фруктозо - 1 - фосфатальдолази , що веде до надмірного накопичення фруктозо - 1 - фосфату в печінці , кишечнику , нирках.
Хвороба проявляється в ранньому віці при вживанні продуктів, що містять плодовий цукор (солодкі фрукти , ягоди , мед , тростинний і буряковий цукор ) і характеризується анорексією , блювотою , гіпоглікемією , гепатомегалією , рідше - асцитом , жовтяницею. Причинами смерті можуть стати гіпоглікемія і ацидоз , прогресуюча кахексія . Гепатоцелюлярні порушення можуть викликати розвиток цирозу печінки і печінкової недостатності. Своєчасне призначення дієти , що не містить фруктози , може призупинити розвиток хвороби.
♦ Сукрозія-
♦ Синдром Марфана
 –аутосомно-домінантне генетичне захворювання сполучної тканини, яке характеризується диспропорційними довгими кінцівками, тонкими худими пальцями, відповідно тонкою статурою та наявністю серцево-судинних вад, що специфічно проявляються у вигляді вад серцевих клапанів та аорти. Синдром Марфана розвивається внаслідок розладу експресії гену FBN1, що розташований на 15-й хромосомі.
–аутосомно-домінантне генетичне захворювання сполучної тканини, яке характеризується диспропорційними довгими кінцівками, тонкими худими пальцями, відповідно тонкою статурою та наявністю серцево-судинних вад, що специфічно проявляються у вигляді вад серцевих клапанів та аорти. Синдром Марфана розвивається внаслідок розладу експресії гену FBN1, що розташований на 15-й хромосомі.
♦ Гаргоілізм
 -рідкісне спадкове захворювання , обумовлене порушенням обміну ліпідів і мукополісахаридів . В результаті обмінних порушень відбувається накопичення в клітинах мозку , сітківці ока , периферичних нервах , печінці, селезінці та інших органах високомолекулярних липоїдно - полісахаридних комплексів .
-рідкісне спадкове захворювання , обумовлене порушенням обміну ліпідів і мукополісахаридів . В результаті обмінних порушень відбувається накопичення в клітинах мозку , сітківці ока , периферичних нервах , печінці, селезінці та інших органах високомолекулярних липоїдно - полісахаридних комплексів .
Типовий зовнішній вигляд дитини: грубі риси обличчя, що навис лоб, запали перенісся, широкі розгорнуті ніздрі, потовщені губ , відкритий рот, великий язик, деформовані вушні раковини. Тулуб укорочено, грудна клітка окреглеа . Можуть визначатися пупкові, пахові- мошоночние грижі; збільшення печінки , селезінки; помутніння рогівки, кіфоз в нижньогрудном відділі хребта; підвищена екскреція мукополісахаридів з сечею.
Лікування: показано застосування гормональних препаратів ( АКТГ, тиреоідина ) за призначенням лікаря.
♦ Синдром Лоуренса
 –характеризується гіпофізарним ожирінням і недорозвиненням статевих ознак у поєднанні з пігментним ретинітом, провідним до сліпоти, недоумством, аномаліями фізичного розвитку (полідактилія, синдактилією, патологічної формою черепа та ін.) Його прийнято розглядати як результат недорозвитку або внутрішньоутробного ураження гіпофізарно-гіпоталамічної системи.
–характеризується гіпофізарним ожирінням і недорозвиненням статевих ознак у поєднанні з пігментним ретинітом, провідним до сліпоти, недоумством, аномаліями фізичного розвитку (полідактилія, синдактилією, патологічної формою черепа та ін.) Його прийнято розглядати як результат недорозвитку або внутрішньоутробного ураження гіпофізарно-гіпоталамічної системи.
♦ Синдром Шерешевського-Тернера
 – захворювання пов'язане з аномалією кількості статевих хромосом. Захворювання характеризується вираженим статевим інфантилізмом, низькорослістю, утворенням типових складок на бічних поверхнях шиї. Низькорослість хворих помірна, у них широка грудна клітка і велика відстань між сосками. Ріст дітей значно уповільнений, кінцевий зріст рідко перевищує 135 см. У хворих можливі такі аномалії розвитку, як деформації вушних раковин і їх низьке розташування, зменшена нижня щелепа, епікантус (третя повіка), полідактилія, синдактилія та ін. Досить часто при цьому синдромі є вади розвитку внутрішніх органів: коярктація аорти, відкрита артеріальна протока, декстракардія, аномалії нирок. Розумовий розвиток переважно нормальний. Найбільш постійним симптомом є дисгенезія гонад. У пацієнток немає яєчників, вони замінені рудиментарними тяжами зі сполучної тканини. Порушення розвитку статевої системи проявляються інфантилізмом. Такі хворі, як правило, неплідні, у них слабко розвинені вторинні статеві ознаки, немає менархе. Особливістю проявів хвороби у дітей грудного віку є наявність лімфатичного набряку стоп, гомілок і кистей, а також крилоподібних складок на шиї.
– захворювання пов'язане з аномалією кількості статевих хромосом. Захворювання характеризується вираженим статевим інфантилізмом, низькорослістю, утворенням типових складок на бічних поверхнях шиї. Низькорослість хворих помірна, у них широка грудна клітка і велика відстань між сосками. Ріст дітей значно уповільнений, кінцевий зріст рідко перевищує 135 см. У хворих можливі такі аномалії розвитку, як деформації вушних раковин і їх низьке розташування, зменшена нижня щелепа, епікантус (третя повіка), полідактилія, синдактилія та ін. Досить часто при цьому синдромі є вади розвитку внутрішніх органів: коярктація аорти, відкрита артеріальна протока, декстракардія, аномалії нирок. Розумовий розвиток переважно нормальний. Найбільш постійним симптомом є дисгенезія гонад. У пацієнток немає яєчників, вони замінені рудиментарними тяжами зі сполучної тканини. Порушення розвитку статевої системи проявляються інфантилізмом. Такі хворі, як правило, неплідні, у них слабко розвинені вторинні статеві ознаки, немає менархе. Особливістю проявів хвороби у дітей грудного віку є наявність лімфатичного набряку стоп, гомілок і кистей, а також крилоподібних складок на шиї.
♦ Синдром Клайнфельтера
 –це найбільш поширене захворювання статевих хромосом у чоловіків і другий найбільш поширений розлад, викликаний наявністю додаткових хромосом. Це порушення виникає приблизно у 1 чоловіка з 1000. Кожен з 500 чоловіків має додаткову Х хромосому, але жодних ознак чи симптомів захворювання у них немає. Синдром XXY може також виникати й у інших ссавці, в т.ч. у мишей. Основним наслідком виникнення захворювання є розвиток маленьких яєчок та зниження репродуктивної здатності. Є ще багато інших типових фізичних і поведінкових відхилень, пов’язаних із синдромом, проте процес перебігу захворювання в кожному конкретному випадку відрізняється. Є багато випадків при яких у чоловіків чи хлопчиків немає жодних видимих ознак хвороби. Уражені цим розладом чоловіки майже завжди безплідні, хоча при використанні новітніх репродуктивних технологій, іноді ситуацію вдається виправити. У деяких випадках можуть виникати проблеми з вивченням мови, а нейропсихологічне тестування часто виявляє порушення виконавчих функцій (в нейропсихології, гіпотетичний набір високорівневих процесів, які дозволяють планувати поточні дії відповідно зі спільною метою, змінювати реакцію в залежності від контексту, вибірково приділяти увагу потрібним стимулам). Більш важкі наслідки цього захворювання пов’язані з підвищеним ризиком виникнення ракових пухлин статевих клітин, чоловічого раку молочної залози та остеопорозу, тобто підвищується ймовірність виникнення тих хвороб, які більше характерні для жінок Крім того, згідно з медичними дослідженнями синдрому Клайнфельтера, він є пов'язаний також з іншими розладами, як хвороби легенів, варикозне розширення вен, цукровий діабет і ревматоїдний артрит, проте ці зв’язки поки що не дуже добре зрозумілі та описані.
–це найбільш поширене захворювання статевих хромосом у чоловіків і другий найбільш поширений розлад, викликаний наявністю додаткових хромосом. Це порушення виникає приблизно у 1 чоловіка з 1000. Кожен з 500 чоловіків має додаткову Х хромосому, але жодних ознак чи симптомів захворювання у них немає. Синдром XXY може також виникати й у інших ссавці, в т.ч. у мишей. Основним наслідком виникнення захворювання є розвиток маленьких яєчок та зниження репродуктивної здатності. Є ще багато інших типових фізичних і поведінкових відхилень, пов’язаних із синдромом, проте процес перебігу захворювання в кожному конкретному випадку відрізняється. Є багато випадків при яких у чоловіків чи хлопчиків немає жодних видимих ознак хвороби. Уражені цим розладом чоловіки майже завжди безплідні, хоча при використанні новітніх репродуктивних технологій, іноді ситуацію вдається виправити. У деяких випадках можуть виникати проблеми з вивченням мови, а нейропсихологічне тестування часто виявляє порушення виконавчих функцій (в нейропсихології, гіпотетичний набір високорівневих процесів, які дозволяють планувати поточні дії відповідно зі спільною метою, змінювати реакцію в залежності від контексту, вибірково приділяти увагу потрібним стимулам). Більш важкі наслідки цього захворювання пов’язані з підвищеним ризиком виникнення ракових пухлин статевих клітин, чоловічого раку молочної залози та остеопорозу, тобто підвищується ймовірність виникнення тих хвороб, які більше характерні для жінок Крім того, згідно з медичними дослідженнями синдрому Клайнфельтера, він є пов'язаний також з іншими розладами, як хвороби легенів, варикозне розширення вен, цукровий діабет і ревматоїдний артрит, проте ці зв’язки поки що не дуже добре зрозумілі та описані.
♦ Синдром Штурге-Вебера-Краббе –генетична , імовірно , патологія. Характерні різні психічні розлади, симптоми когнітивного дефіциту. Спостерігаються також плоска гемангіома особи в зоні іннервації трійчастого нерва на одній стороні особи, ангіома на очному дні і глаукома на тій же стороні. Часто виявляється гемангіома менингиальной оболонки і пов'язані з нею епілептичні припадки , а також осередкові звапніння в мозку, іноді асиметрія кісток обличчя і черепа (атрофія кісток на одній стороні). Лікування симптоматична, ефективних методів профілактики нині не існує.
♦ Дисплазія –загальна назва наслідків неправильного формування у процесі ембріогенезу та постнатальному періоді окремих частин, органів або тканин організму; зміна розміру, форми та будови клітин, тканин або органів. Відбувається поширення недорозвинених клітин, з відповідним зменшенням кількості та зміною місцезнаходження нормальних клітин. Дисплазія часто є показником початку неопластичних процессів.
Дисплазія характеризується чотирма головними патологічними змінами:
1. Анізоцитоз (клітини нерівні за розміром);
2. Пойкілоцитоз (клітини аномальної форми);
3. Гіперхроматоз (аномалії в пігментації);
4. Аномалії у мітотичній активності клітин.
Дисплазія, при якій розвиток та диференціація клітин не відбуваються, може бути протиставлена метаплазії, при якій клітини одного типу заміщуються клітинами іншого.
♦ Арахнодaктилія («павукові пальці»)
 -це патологічний стан, при якому пальці аномально подовжені і вузькі в порівнянні з долонею. Ця хвороба може бути вродженою чи набутою протягом життя. Слід зазначити, що в деяких випадках арахнодактиліі всі або кілька пальців на руці володіють великою гнучкістю і здатністю бути відхиленими назад на 180 °.
-це патологічний стан, при якому пальці аномально подовжені і вузькі в порівнянні з долонею. Ця хвороба може бути вродженою чи набутою протягом життя. Слід зазначити, що в деяких випадках арахнодактиліі всі або кілька пальців на руці володіють великою гнучкістю і здатністю бути відхиленими назад на 180 °.
Подобное состояние может проявиться само по себе, без каких-либо сопутствующих проблем со здоровьем. Тем не менее, оно зачастую связано с определёнными группами заболеваний, таких как синдром Марфана (при этом заболевании проявляются близорукость (после 10-15 лет), высокая подвижность суставов, не пропадающая с возрастом, длинные конечности. Часто больные худощавы и высокорослы).
Арахнодактилию принято связывать с мутациями в обоих генах фибриллин-1 и фибриллин-2.
♦ Іхтіоз
 –це група спадкових захворювань шкіри, яка характеризується порушеннями зроговіння. Етіологія невідома. Розрізняють декілька клінічних форм, обумовлених різними групами мутантних генів, біохімічний дефект яких остаточно не розшифрований. Надають велике значення недостатності вітаміну А, ендокринопатії (гіпофункції щитовидної залози, статевих залоз). Патологічний процес - гіперкератоз - призводить до появи на шкірі лусочок, що нагадують риб'ячу луску.
–це група спадкових захворювань шкіри, яка характеризується порушеннями зроговіння. Етіологія невідома. Розрізняють декілька клінічних форм, обумовлених різними групами мутантних генів, біохімічний дефект яких остаточно не розшифрований. Надають велике значення недостатності вітаміну А, ендокринопатії (гіпофункції щитовидної залози, статевих залоз). Патологічний процес - гіперкератоз - призводить до появи на шкірі лусочок, що нагадують риб'ячу луску.
♦ Синдром котячого лементу (крику; Синдром Лежена)
 –рідкісне генетичне захворювання, що пов'язане з відсутністю частини 5 хромосоми. Уражені цим захворюванням діти (переважно, але не можна сказати, що усі діти) мають плач, який схожий на котячий крик, саме тому цей синдром отримав назву від фр. Cri-Du-Chat Syndrome, що дослівно озаначає «плач кішки або крик кота». Частота виникнення синдрому — 1 дитина на 50000 народжених, зустрічається у всіх етнічних груп та частіше нею хворіють жінки, співвідношення чоловічої і жіночої статті становить 4:3.
–рідкісне генетичне захворювання, що пов'язане з відсутністю частини 5 хромосоми. Уражені цим захворюванням діти (переважно, але не можна сказати, що усі діти) мають плач, який схожий на котячий крик, саме тому цей синдром отримав назву від фр. Cri-Du-Chat Syndrome, що дослівно озаначає «плач кішки або крик кота». Частота виникнення синдрому — 1 дитина на 50000 народжених, зустрічається у всіх етнічних груп та частіше нею хворіють жінки, співвідношення чоловічої і жіночої статті становить 4:3.
Як вже було зазначено, синдром одержав свою назву через характерний плач дітей (він аналогічний м’явканню кошеняти, крику кішки), які страждають від цього захворювання. Це явище відбувається через проблеми з гортанню і нервовою системою. Близько 1/3 дітей втрачають цю особливу характерну рису до 2 років. Іншими симптомами, які вказують на захворювання синдромом котячого крику є:
- проблеми з харчуванням через труднощі при ковтанні і смоктанні;
- низька вага при народженні та низькі темпи розвитку (в першу чергу фізичного);
- суттєва затримка розвитку когнітивних, мовленнєвих функцій та функцій руху;
- поведінкові проблеми, такі як гіперактивність, агресія, істерики і одноманітні рухи, які постійно повторюються;
- нетипові риси обличчя, які можуть з часом зникнути або посилитися;
- надмірне, неконтрольоване слиновиділення;
- закрепи.
♦ Синдром де Груши
 –синдром пов'язаний з повною або частковою делецією(структурна мутація) короткого плеча хромосоми 18. Від нормальної тривалості вагітності діти народжуються з невеликою масою тіла ( в середньому 2800 хворі маленького зросту, з мікроцефальним черепом. Круглим обличчям і вогнищами облисіння на голові. Очні щілини горизонтальні, широке перенісся. Птоз або косоокість. Верхня губа коротка і виступає вперед , у формі " поліцейської кашкети" . Підборіддя, виступаюче у деяких новонароджених, з віком приходить до норми, зуби майже завжди каріозні, нерідко відсутні латеральні різці, описано кілька хворих з одним центральним різцем на верхній щелепі. Вуха великі, відстовбурчені, деформовані, з недорозвиненим завитка, розташовані низько і відсунуті назад. Шия коротка, іноді з крилоподібні складкою і низьким зростанням волосся. Грудна клітка вкорочена, соски широко розставлені; діастаз прямих м'язів живота пупкові і пахові грижі свідчать про м'язової гіпотонії. Кисті широкі і короткі, кінцеві фаланги звужені, відзначаються клинодактилія у пальців, сіндактілія пальців ніг і деформація стоп. Набряклість тильної поверхні кистей доповнюють загальну картину і роблять її схожою на фенотип при синдромі Шерешевського - Тернера. У хлопчиків знаходять ектопію яєчок, гіпоспадію, недорозвинення статевого члена.
–синдром пов'язаний з повною або частковою делецією(структурна мутація) короткого плеча хромосоми 18. Від нормальної тривалості вагітності діти народжуються з невеликою масою тіла ( в середньому 2800 хворі маленького зросту, з мікроцефальним черепом. Круглим обличчям і вогнищами облисіння на голові. Очні щілини горизонтальні, широке перенісся. Птоз або косоокість. Верхня губа коротка і виступає вперед , у формі " поліцейської кашкети" . Підборіддя, виступаюче у деяких новонароджених, з віком приходить до норми, зуби майже завжди каріозні, нерідко відсутні латеральні різці, описано кілька хворих з одним центральним різцем на верхній щелепі. Вуха великі, відстовбурчені, деформовані, з недорозвиненим завитка, розташовані низько і відсунуті назад. Шия коротка, іноді з крилоподібні складкою і низьким зростанням волосся. Грудна клітка вкорочена, соски широко розставлені; діастаз прямих м'язів живота пупкові і пахові грижі свідчать про м'язової гіпотонії. Кисті широкі і короткі, кінцеві фаланги звужені, відзначаються клинодактилія у пальців, сіндактілія пальців ніг і деформація стоп. Набряклість тильної поверхні кистей доповнюють загальну картину і роблять її схожою на фенотип при синдромі Шерешевського - Тернера. У хлопчиків знаходять ектопію яєчок, гіпоспадію, недорозвинення статевого члена.
Практично у всіх випадках виявляється розумова відсталість від легкої дебільності до ідіотії. Майже всі діти відстають у моторному і фізичному розвитку, у ряді випадків відзначається судомний синдром. Для цієї патології типові гіпомімія і більш виражені порушення експресивної мови в порівнянні з глибиною інтелектуального дефекту. Таку дисоціацію М. Г. Блюміна , О. А. Подугольнікова ( 1969 ) пояснюють поєднанням дизартрії з грубим недорозвиненням корковою моторної функції мови .
♦ Синдром Патау
 -генетичне захворювання, хромосомна аномалія, синдром, при якому пацієнт має додаткову 13 хромосому.
-генетичне захворювання, хромосомна аномалія, синдром, при якому пацієнт має додаткову 13 хромосому.
При синдромі Патау спостерігаються важкі вроджені пороки. Діти з синдромом Патау народжуються з масою тіла нижче норми (2500 г). У них виявляються помірна мікроцефалія, порушення розвитку різних відділів ЦНС, низький скошений лоб, звужені очні щілини, відстань між якими зменшено, микрофтальмия і колобома, помутніння рогівки, запали перенісся, широке підставу носа , деформовані вушні раковини, ущелина верхньої губи і піднебіння , полідактилія , флексорного положення кистей, коротка шия. У 80% новонароджених зустрічаються вади розвитку серця: дефекти міжшлуночкової та міжпередсердної перегородок, транспозиції судин та ін Спостерігаються фіброкистозною зміни підшлункової залози, додаткові селезінки , ембріональна пупкова грижа. Нирки збільшені, мають підвищену дольчатість і кісти в кірковому шарі, виявляються вади розвитку статевих органів. Для СП характерна затримка розумового розвитку.
У зв'язку з важкими вродженими вадами розвитку більшість дітей з синдромом Патау помирають у перші тижні або місяці ( 95 % - до 1 року). Однак деякі хворі живуть протягом кількох років.
Виправити хромосомні порушення неможливо. Комплексна робота групи різних фахівців полягає в постійному контролі за станом здоров'я хворого і підтримці сім'ї.
♦ Синдром Реторе
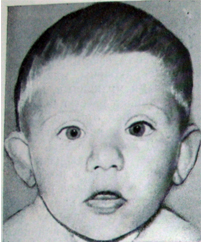 –цитогенетичнп основа синдрому є тріплікація коротких плечей хромосоми 9 (у кількох хворих тетраплікація), а в деяких випадках і тріплікаціі невеликої ділянки довгих плечей, то в клінічній картині синдрому можна побачити частину симптомів, характерних для повної трисомії 9. Череп у новонароджених мікробрахіцефальний з плоским потилицею, виступаючим лобом, широко відкритими тім'ячками і лобовим швом ; з віком брахіцефалія зменшується. Очні щілини маленькі, антімонголоідний розрізу, очні яблука посаджені глибоко, гипертелоризм, епікант , косоокість, ніс великий, "м'ясисти ", особливо кінчик, спинка носа виступає в середній частині, ніздрі відкриті донизу, перегородка виходить за площину ніздрів; верхня губа вкорочена, нижня кілька вивернута, кути рота опущені, рот відкривається асиметрично. Великі відстовбурчені вушні раковини розташовані нормально. На думку деяких клініцистій, підняті брови, косий розріз очей і опущені кути рота надають особі вираз заклопотаності, тривожності.
–цитогенетичнп основа синдрому є тріплікація коротких плечей хромосоми 9 (у кількох хворих тетраплікація), а в деяких випадках і тріплікаціі невеликої ділянки довгих плечей, то в клінічній картині синдрому можна побачити частину симптомів, характерних для повної трисомії 9. Череп у новонароджених мікробрахіцефальний з плоским потилицею, виступаючим лобом, широко відкритими тім'ячками і лобовим швом ; з віком брахіцефалія зменшується. Очні щілини маленькі, антімонголоідний розрізу, очні яблука посаджені глибоко, гипертелоризм, епікант , косоокість, ніс великий, "м'ясисти ", особливо кінчик, спинка носа виступає в середній частині, ніздрі відкриті донизу, перегородка виходить за площину ніздрів; верхня губа вкорочена, нижня кілька вивернута, кути рота опущені, рот відкривається асиметрично. Великі відстовбурчені вушні раковини розташовані нормально. На думку деяких клініцистій, підняті брови, косий розріз очей і опущені кути рота надають особі вираз заклопотаності, тривожності.
Шия коротка, з натяком на шкірну складку, грудна клітка широка, із збільшеною відстанню між сосками. Нерідко спостерігаються розбіжність прямих м'язів живота, грижі пупкового кільця і білої лінії, світлі плями на шкірі і смуги на животі, виражені западини під ключицями і в області крижів. У деяких хворих порушена постава.
♦ Синдром Ретта
 –психоневрологічне спадкове захворювання, зустрічається майже виключно у дівчат з частотою 1:10000 - 1:15000, спричиняє важку розумову відсталість у дівчат. Характерним для даного стану є стереотипні, одноманітні рухи рук, їх потирання, заламування, які при цьому не носять цілеспрямованого характеру. Мова утруднюється, відповіді стають одноманітними або ехолалічними, часом мова зовсім пропадає (мутизм). Спостерігається низький психологічний тонус. Обличчя дитини поступово набуває сумного, «неживого» виразу, погляд стає розфокусованим або спрямованим в одну точку перед собою. Рухи стають загальмованими, але можливі напади насильницького сміху разом з нападами імпульсивної поведінки. З'являються судомні припадки. Ці особливості нагадують поведінку дітей з раннім дитячим аутизмом.
–психоневрологічне спадкове захворювання, зустрічається майже виключно у дівчат з частотою 1:10000 - 1:15000, спричиняє важку розумову відсталість у дівчат. Характерним для даного стану є стереотипні, одноманітні рухи рук, їх потирання, заламування, які при цьому не носять цілеспрямованого характеру. Мова утруднюється, відповіді стають одноманітними або ехолалічними, часом мова зовсім пропадає (мутизм). Спостерігається низький психологічний тонус. Обличчя дитини поступово набуває сумного, «неживого» виразу, погляд стає розфокусованим або спрямованим в одну точку перед собою. Рухи стають загальмованими, але можливі напади насильницького сміху разом з нападами імпульсивної поведінки. З'являються судомні припадки. Ці особливості нагадують поведінку дітей з раннім дитячим аутизмом.
♦ Синдром Нунан
 – рідкісна вроджена патологія, успадковується за аутосомно-домінантним типом, носить сімейний характер, проте зустрічаються і спорадично. Синдром припускає наявність фенотипу, характерного для синдрому Шерешевського-Тернера у особин жіночої та чоловічої статі з нормальним генотипом.
– рідкісна вроджена патологія, успадковується за аутосомно-домінантним типом, носить сімейний характер, проте зустрічаються і спорадично. Синдром припускає наявність фенотипу, характерного для синдрому Шерешевського-Тернера у особин жіночої та чоловічої статі з нормальним генотипом.
Характеризується низькорослістю (кінцевий зростання у хлопчиків досягає 162 см , у дівчаток - 153 см ) і гіпогонадизмом . Недостатність гонад різна:
-у чоловіків - від повної агенезії до невеликої гіпоплазії яєчок - яєчка маленькі , часто відзначається крипторхізм , сперматогенез відсутня або виявляється різна ступінь олігозооспермії . Слабо розвинені вторинні статеві ознаки ( убоге статеве оволосіння , слабо розвинена мускулатура ) ;
-у жінок часто функція яєчників нормальна , можуть бути менструації , можлива навіть фертильність. Відзначається гіпоплазія зовнішніх геніталій .
Симптоматика:
-основні клінічні прояви даного синдрому мають схожість з синдромом Шерешевського - Тернера : крилоподібні складки на шиї , вальгусна деформація ліктьових суглобів , низькорослість , лімфатичні набряки кистей і стоп;
інші прояви синдрому: птоз , запала грудна клітка , вроджені вади правої половини серця -(стеноз легеневої артерії) , трикутне обличчя і розумова відсталість. У хлопчиків відзначаються порушення розвитку яєчок ( крипторхізм , атрофія , анорхія , зменшення просвіту сім'явивідних канальців зі склерозом або без нього , зменшення або відсутність гермінальних клітин , гіперплазія клітин Лейдіга ) і мікропенія . Деякі пацієнти з нормальними яєчками зберігають фертильність , проте у більшості відзначається помірний або виражений гіпогонадизм . Вміст тестостерону в плазмі крові низький або визначається на нижніх межах норми , рівень гонадотропінів підвищений. Каріотип XY ( нормальна, чоловіча ) . Причина затримки зростання не уточнена , так як рівень базального та стимульованого гормону росту нормальний . Зміст ІФР - 1 знижене або залишається на нижніх межах норми .
♦ Синдром Рубінштейна-Тейбі
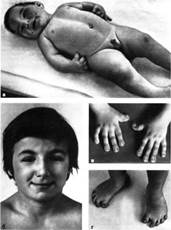 –це специфічний розлад, при якому певні фізичні ознаки та аномалії розвитку виникають разом. Особи із цим синдромом зазвичай невисокі на зріст, відстають у розвитку, мають характерні риси обличчя та широкі перші пальці рук та ніг. Вважається, що цей синдром зустрічається з частотою 1 на 30000 народжень. Однаково часто хворіють дівчатка та хлопчики. Більшість дітей із синдромом Р-Т при народженні не схожі на своїх батьків. Встановити синдром при народженні дуже важко (середній вік встановлення діагнозу – 15 місяців), іноді він не проявляється протягом багатьох років. На жаль, не існує медичних тестів, щоб допомогти встановити синдром Р-Т. Єдиний можливий спосіб – помітити специфічні риси. Характерними рисами обличчя є – маленька голова, низький ріст волосся (волосся жорстке та іноді росте низько на лобі), антимонголоїдний розріз очей, гострий ніс, маленький рот та високе піднебіння. Специфічними є також широкі великі пальці на руках та ногах. Більшість цих ознак важко розпізнати настільки, щоб це було підставою для встановлення діагнозу. Ці риси можуть бути непомітними для родичів та друзів, але дозволять спеціалісту-генетику або педіатру діагностувати синдром Р-Т. Причини виникнення синдрому Р-Т невідомі. При цьому синдромі не встановлено ніяких аномалій хромосом. Однак, не виключається мутація гена. Не підтверджено також вплив хімічних чи інших речовин під час вагітності, що можуть бути причиною виникнення синдрому. Генетична природа синдрому Р-Т не підтверджена. У більшості випадків, синдром зустрічається лише у однієї дитини певної родини. У медичній літературі згадувалось про сім сімейних випадків синдрому Р-Т, однак більшість з них були описані недостатньо точно. Серед них згадувались два ймовірні випадки синдрому Р-Т, де брат та сестра у одній родині мали цей синдром, а у іншій – синдром діагностували у матері та сина. З огляду на це, ризик народження ще однієї дитини із синдромом у родині становить близько 1 відсотка, тоді як ризик народження дитини особою, якій діагностували цей синдром, складає 50 відсотків. Методи пренатальної діагностики синдрому Р-Т не розроблені.
–це специфічний розлад, при якому певні фізичні ознаки та аномалії розвитку виникають разом. Особи із цим синдромом зазвичай невисокі на зріст, відстають у розвитку, мають характерні риси обличчя та широкі перші пальці рук та ніг. Вважається, що цей синдром зустрічається з частотою 1 на 30000 народжень. Однаково часто хворіють дівчатка та хлопчики. Більшість дітей із синдромом Р-Т при народженні не схожі на своїх батьків. Встановити синдром при народженні дуже важко (середній вік встановлення діагнозу – 15 місяців), іноді він не проявляється протягом багатьох років. На жаль, не існує медичних тестів, щоб допомогти встановити синдром Р-Т. Єдиний можливий спосіб – помітити специфічні риси. Характерними рисами обличчя є – маленька голова, низький ріст волосся (волосся жорстке та іноді росте низько на лобі), антимонголоїдний розріз очей, гострий ніс, маленький рот та високе піднебіння. Специфічними є також широкі великі пальці на руках та ногах. Більшість цих ознак важко розпізнати настільки, щоб це було підставою для встановлення діагнозу. Ці риси можуть бути непомітними для родичів та друзів, але дозволять спеціалісту-генетику або педіатру діагностувати синдром Р-Т. Причини виникнення синдрому Р-Т невідомі. При цьому синдромі не встановлено ніяких аномалій хромосом. Однак, не виключається мутація гена. Не підтверджено також вплив хімічних чи інших речовин під час вагітності, що можуть бути причиною виникнення синдрому. Генетична природа синдрому Р-Т не підтверджена. У більшості випадків, синдром зустрічається лише у однієї дитини певної родини. У медичній літературі згадувалось про сім сімейних випадків синдрому Р-Т, однак більшість з них були описані недостатньо точно. Серед них згадувались два ймовірні випадки синдрому Р-Т, де брат та сестра у одній родині мали цей синдром, а у іншій – синдром діагностували у матері та сина. З огляду на це, ризик народження ще однієї дитини із синдромом у родині становить близько 1 відсотка, тоді як ризик народження дитини особою, якій діагностували цей синдром, складає 50 відсотків. Методи пренатальної діагностики синдрому Р-Т не розроблені.
♦ Синдром Вільямса
 –синдром, що виникає унаслідок хромосомної патології. Люди, що страждають ним мають специфічну зовнішність і характеризуються загальною затримкою розумового розвитку, дуже низьким IQ, побутовою непристосованістю, при цьому мають вільну дуже розвинену мову з використанням рідкісних слів.
–синдром, що виникає унаслідок хромосомної патології. Люди, що страждають ним мають специфічну зовнішність і характеризуються загальною затримкою розумового розвитку, дуже низьким IQ, побутовою непристосованістю, при цьому мають вільну дуже розвинену мову з використанням рідкісних слів.
Хворі мають особливу будову обличчя, у спеціальній літературі зване «обличчям ельфа», оскільки воно нагадує обличчя ельфів в їх традиційному, фольклорному варіанті. Для них характерні широкий лоб, розліт брів по середній лінії, опущені вниз повні щоки, великий рот з повними губами (особливо нижня), пласке перенісся, своєрідна форма носа з пласким тупим кінцем, маленьке, трохи загострене підборіддя.
Очі часто яскраво-блакитні зі зірчастою картиною райдужки та склерами синюватого кольору. Розріз очей своєрідний, з припухлостями навколо повік.
Для старших дітей характерні довгі, рідкі зуби.
Подібність осіб посилює посмішка, яка ще більше підкреслює набряклість повік і своєрідну будову рота.
Жодна з цих рис не є обов'язковою, але їх загальне поєднання завжди присутнє.
Для цього синдрому характерний дефіцит наочно-образного мислення. Розумові порушення спостерігаються також в вербальних здібностях.
ПРИЧИНИ:
Рідкісна делеція (випадання) 26 генів на сьомій хромосомі, клінічно проявляється у формі гіперкапнії.
1) Особливості морфології мозку;
Головний мозок – це орган, який відіграє найбільш важливу роль у життєдіяльності людини, це орган пристосування до умов оточуючого середовища. Маса головного мозку складає 10 % маси тіла новонародженого (приблизно 350-400 г), в дітей віком 16-17 років - 2,5 % від маси тіла (близько 1300-1400 г). До 2-х років маса мозку дитини дорівнює масі мозку дорослого.Окремі частки мозку розвиваються нерівномірно (лобна і тім’яна долі ростуть швидше скроневої і потиличної). У новонароджених і дітей дошкільного віку головний мозок коротший і ширший. До 4 років ріст мозку у довжину, ширину і висоту майже рівномірний, а з 4 до 7 років найбільш інтенсивно збільшується його висота.
Морфологічні особливості головного мозку новонароджених дітей:
- мозкова тканина багата водою (91,5%);
- містить мало лецитину та інших білкових речовин, відрізняється хімічною будовою нейроглобуліну інейростроміну;
- після народження продовжується зміна форми і величини борозн і звивин, особливо енергійно вони проходять у перші 5 років життя;
- число клітин макроглії в півкулях головного мозку новонародженого таке ж, як у дорослого, але вони ще не зрілі;
- дозрівання клітин кори закінчується до 18-20 місяців, вони не мають відростків (аксонів і дендритів).