|
|
Химические свойства алканов
Высокая прочность С-С и С-Н связей в алканах обусловливают их высокую инертность, а низкая полярность и поляризуемость – склонность к гомолитическим реакциям. По этим причинам большинство реакций алканов имеет радикальный характер, а для успеха их протекания требуется внешнее энергетическое воздействие (например, температура, фотолиз, катализ).
Свободнорадикальные реакции алканов
1. Галогенирование.
Алканы очень активно взаимодействуют с фтором, реакция с хлором происходит при освещении и нагревании, взаимодействие с бромом осуществляют при совместном освещении и нагревании. Такие различия обусловлены различной энергетикой реакций галогенирования.
Механизм развития цепей этих реакций можно представить двумя последовательными стадиями.
X· + RH ® HX + R· (1)
R· + X2 ® RX + X· (2)
При достаточно длинных кинетических цепях стехиометрия брутто-реакции определяется реакциями (1) и (2). Суммирование левых и правых частей уравнений этих реакций приводит к стехиометрическому уравнению.
RH + X2 ® RX + HX
Фторирование алканов
В случае фторирования реакция протекает самопроизвольно со взрывом из-за высокого теплового эффекта каждой стадии и быстроты их протекания. Инициирование осуществляется посредством бимолекулярной реакции
F2 + RH ® RF + F· + R·
эндотермичность которой не превышает 20 кДж/моль. Это обеспечивает умеренное cкорoсти инициирования. При этом высокая экзотермичность реакций (1) и (2) обусловливает близкие к нулю их энергии активации и высокие скорости. В целом это приводит к столь высоким скоростям брутто-реакции и соответственно скоростям тепловыделений, что большая часть выделяющегося тепла идет на разогрев реакционной смеси, приводящего в конечном счете к тепловому взрыву. На практике, чтобы осуществить фторирование с контролируемыми скоростями исходные реагенты разбавляют инертными газами (N2, Ar и др).
Хлорирование алканов
Расщепление молекулы хлора на хлоррадикалы
Cl2 + M ® M + 2Cl·
требует подвода энергии, равной энергии связи Cl-Cl 243 кДж/моль. Поэтому в обычным условиях реакция хлорирования не идет. Более эффективное инициирование можно осуществить путем.:
а) Повышения температуры
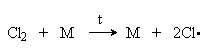
б) Фотооблучения реакционной смеси УФ-светом.
Cl2 + hn ® 2Cl·
в) Проведения реакции в присутствии иницииаторов – соединений, легко диссоциирующих на свободные радикалы, которые дают начало цепному процессу.
In2 ® 2In·
In· + Cl2 ® InCl + Cl·
В качестве инициаторов хлорирования и других свободно радикальных реакций можно использовать органические пероксиды и азосоединения, например бензоилпероксид

или азобисизобутиронитрил
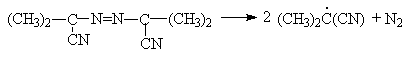
Любой из способов инициирования более эффективно генерирует свободные радикалы, благодаря чему их концентрация становится достаточной, чтобы обеспечить высокие скорости брутто-реакций. Реакции развития цепей (1) и (2) в случае хлорирования метана имеют тепловые эффекты соответственно –4,2 и –109 кДж/моль. Экзотермичность обоих стадий обусловливает низкие энергетические барьеры реакции (1) и (2), что при эффективном инициировании обеспечивает достаточно высокие скорости реакций хлорирования.
Бромирование алканов
Стадия развития цепей (1) для реакции бромирования метана умеренно эндотермична (+67 кДж/моль). Это обусловливает ее высокий энергетический барьер и обратимость. Как следствие длины кинетических цепей низки (от нескольких единиц до 2-3 десятков). Поэтому бромирование метана и других мало реакционноспособных субстратов протекает трудно, более эффективно реакция протекает с реакционноспособными субстратами.
Иодирование алканов
Реакция иодирования алканов оказывается невозможной, так как стадия (1) высоко эндотермична (+138 кДж/моль). Это обусловливает смещение равновесия этой реакции нацело в левую сторону. Наоборот, известно, что йодалканы под действием НJ легко превращаются в алканы и йод.
Наибольший практический интерес имеют реакция хлорирования алканов и их производных.
Региоселективностьреакций свободнорадикального галогенирования
При свободнорадикальном галогенировании сложного алкана, имеющего в своей структуре различные связи С-Н, образуются смеси различных галогеналканов. При этом состав образующейся смеси определяется относительной реакционной способностью различных связей С-Н в реакции отрыва водорода (1) и их числом.
Ряд относительных реакционных способностей связей С-Н в реакциях радикального замещения.
(C-H)трет > (C-H)втор > (C-H)перв
определяется рядом стабильности образующихся при отрыве водорода в реакции (1) радикалов
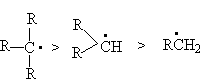
Последний вытекает из действия на стабильность радикалов двух структурных факторов: индуктивного эффекта алкильных групп и эффекта снятия стерических напряжений.
Таким образом, относительные реакционные способности связей С-Н и их число определяют конкуренцию реакций отрыва водорода радикалами Х· от различных положений алканов, а, следовательно, региоселективность радикального замещения.
Задача:
Рассчитайте состав продуктов свободнорадикального хлорирования 2,3-диметилбутана, если реакционные способности третичной и первичной связей С-Н относятся как 4:1
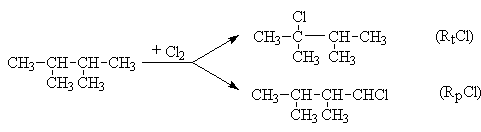
Относительная скорость хлорирования третичных связей С-Н равна произведению числа этих связей на их относительную реакционную способность т.е.
rt=nt.Rt=2.4=8
Соответственно относительная скорость хлорирования первичных связей С-Н равна
rp=np.Rp=12.1=12
Очевидно, что соотношение третичного хлор алкана к первичному равна соотношению скоростей их образования
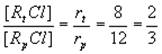
В сумме количества RtCl и RpCl составляет 2+3=5. Тогда доля третичного хлоралкана составляют
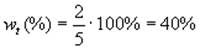
а доля первичного
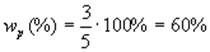
2. Сульфохлорирование
RH + SO2 + Cl2 ® RSO2Cl + HCl
Сульфохлорирование – типичный свободно радикальный процесс. Реакция может инициироваться УФ-светом или химическими инициаторами.
Инициирование:

Развитие цепей
Cl§ +RH ® HCl + R§
R§ +SO2 ® RS§O2
RS§O2 + Cl2® RSO2Cl + Cl§
Обрывцепей
Cl § + Cl § + M ® Cl2 + M
R § + Cl§ ® RCl
R § + R§ ® R-R
Побочно с сульфохлорированием протекает реакция хлорирования:
R § + Cl2 ® RCl + Cl §
Можно видеть, что для подавления побочной реакции необходимо использовать высокие соотношения SO2/Cl2 .Практический интерес представляют реакции сульфохлорирования высших алканов С12 – С18, так как образующиеся сульфохлориды, являются основой для получения ценных неионногенных ПАВ:
RSO2Cl + 2NaOH ® RSO3Na + NaCl + H2O
3. Нитрование алканов
Нитрование алканов – типичная реакция свободнорадикального замещения. В качестве нитрующих агентов выступают оксиды азота или азотная кислота (10-25%)
RH + HONO2 ® RNO2 + H2O (1)
Реакция осуществляется в жидкой фазе при температурах 140-150оС под давлением (реакция Коновалова).
Свободные радикалы образующиеся в результате термического гомолиза азотной кислоты.
HNO3 ® HO· + NO2 (2)
вовлекается в последующие превращения
HO· + RH ® R· + H2O (3)
R· + NO2 ® RNO2 (4)
Суммирование левых и правых частей уравнений (2)-(4) приводит к стехиометрическому уравнению (1).
На самом деле реакция нитрования осложняется деструкцией углеродной цепи с образованием побочных низкомолекулярных нитроалканов и продуктов окисления.
4. Реакции окисления.
Реакции окисления алканов осуществляет в газовой и жидкой фазах. Из газофазных реакций практическое значение имеет окисление метана. Окисление метана имеет последовательно–параллельный характер в соответствии со схемой:

Процесс осуществляют с целью промышленного синтеза метанола и формальдегида. Различают термическое и каталитическое окисление. Первое осуществляют при температурах 500-600о, второе – при 350-450о и катализе AlPO4.
Жидкофазное окисление имеет практическое значение для синтеза высших карбоновых кислот, являющихся сырьем для получения ПАВ:
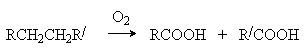
Эти процессы проводят при 120-150оС при инициировании солями Со или Mn, например
Co+3 + RH ® Co+2 + R· + H+
Образующиеся радикалы R· дают начало цепному процессу, который представляет собой совокупность цепей свободнорадикальных реакций с вырожденным разветвлением цепей. Ключевую роль в этом процессе играют образование a -кетогидропероксида, который вовлекается в реакции деструктивного окисления с образованием карбоновых кислот.
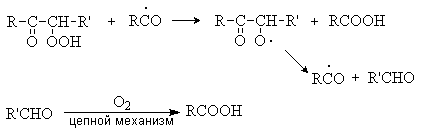
5. Реакции крекинга.
При нагревании выше 500оС алканы подвергаются пиролитическому разложению с образованием сложных смеси продуктов, соотношение которых зависит от температуры и времени реакции. Вся совокупность протекающих при этом реакций называется крекингом. Это типичный свободнорадикальный процесс, инициируемый гомолитическим распадом углерод-углеродных связей исходных алканов. Механизм этого процесса может быть представлен на примере крекинга н-бутана:
Инициирование
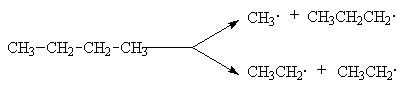
Развитие цепей

В соответствии с приведенным механизмом основными продуктами крекинга являются алканы и алкены, содержащие меньшее число углеродных атомов, чем исходный алкан.
Если крекинг проводить более глубоко и в жестких условиях, то такой исчерпывающий процесс приводит к преимущественному образованию этилена. Этот процесс называется пиролизом. Он реализуется в широких масштабах для производства этилена.
Другим важным процессом превращения алканов является каталитический крекинг, осуществляемый на алюмосиликатных катализаторах при 400-450оС. Изомеризующее и ароматизирующее действие катализаторов в этом процессе обусловливает образование в значительных количествах алканов с разветвленной структурой и арены. Благодаря этому образующиеся углеводородные фракции имеют высокое октановое число. Поэтому каталитическийкрекинг является основным процессом для промышленного производства высокооктановых бензинов.
Особое место среди процессов крекинга занимает термический крекинг метана, осуществляемый для промышленного синтеза ацетилена.
CH4 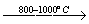 CH2=CH2 + CHº CH + H2
CH2=CH2 + CHº CH + H2
С целью обеспечения необходимой температуры процесса его совмещают с процессом сжигания метана в едином реакционном объеме.
Лекции №11-13
АЛКЕНЫ
План.
- Изомерия и номенклатура алкенов.
- Методы получения.
- Физические и химические свойства.
Алкены (олефины) - углеводороды, в молекулах которых содержится одна двойная углерод-углеродная связь >C=C< между атомами углерода в sp2-состоянии, а все остальные атомы углерода находятся в sp3 - состоянии.
Алкены образуют гомологический ряд соединений, состав которых выражается эмпирической формулой CnH2n.
Изомерияи номенклатура алкенов
Структурная изомерия алкенов начинается с четвертого члена ряда, однако, число структурных изомеров по сравнению с соответствующими алканами значительно больше. Это связано с тем, что структурная изомерия алкенов определяется дополнительным фактором: положением двойной связи углеродной цепи. У алкенов проявляется и другой вид изомерии - пространственная (геометрическая) или цис-транс-изомерия. Так как свободного вращения по двойной связи не происходит, определенные заместители могут фиксироваться по одной стороне двойной связи (цис-изомер), или по противоположным сторонам (транс-изомер):
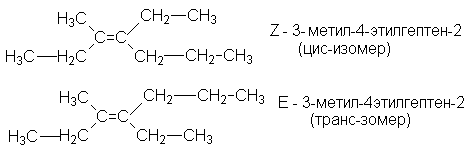
Наличие в молекулах алкенов третичных углеводородных атомов с четырьмя различными заместителями обусловливает их хиральность и, как следствие, наличие стереоизомеров.
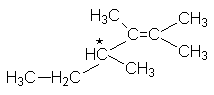
Название алкенов по номенклатуре IUPAC образуются из названий аналогично построенных алканов заменой окончания "ан" на "ен" с указанием цифрой местоположения двойной связи. В качестве родоначальной структуры выбирается самая длинная углеродная цепь, содержащая двойную связь. Нумерация атомов углерода в родоначальной структуре начинается с конца, к которому ближе расположена двойная связь. Иногда употребляется тривиальные и старые рациональные названия алкенов. Так, по рациональной номенклатуре алкены рассматриваются как результат замещения атомов водорода в этилене на алкильные группы.
| Соединения | Названия | |||||
| IUPAC | рациональная | тривиальная | ||||
| СH2=СH2 | этен | этилен | ||||
| CH3CH=CH2 | пропен | метилэтилен | пропилен | |||
| CH2=CH-CH2 -CH3 | 1-бутен | этилэтилен | бутилен | |||
| CH3-CH=CH-CH3 | 2-бутен | симм. диметилэтилен | псевдобутилен | |||
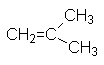
| 2-метилпропен | несимм. диметилэтилен | изобутилен | |||
| CH2=CH-CH2CH2CH3 | 1-пентен | пропилэтилен | амилен | |||
| CH3-CH=CH-CH2-CH3 | 2-пентен | симм. метилэтилен | - | |||
| 3-метил-2-бутен | изопропилэтилен | - | |||
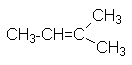
| 2-метил-2бутен | триметилэтилен | - | |||

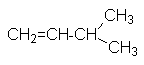
| 2-метил-1-бутен | несимм. метилэтилэтилен | - | |||

| 5-метил-3-изопропил-2-гексен | - | - | |||
Названия остатков алкенов (алкенильных групп) образуют присоединением к названию алкена суффикса "ил". Некоторые остатки алкенов сохраняют тривиальные названия:

Для обозначения геометрических изомеров используется Z, E- номенклатура, согласно которой обозначения Z используется для цис-изомеров, а Е - для транс-изомеров. Обозначения Z и Е- выбираются по пространственному расположению самых старших групп. Старшинство углеводородных заместителей увеличивается в ряду:

Чтобы отнести изомер к Z- или Е- ряду необходимо среди четырех заместителей у двойной связи найти два самых старших. Если оба старших заместителя расположены по одну сторону плоскости, в которой лежит двойная связь, изомер относится к Z-ряду, в противном случае к Е-ряду. (см. пример названий выше).
Методыполучения алкенов
1. Дегидрирование и крекинг алканов.
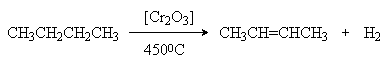
При более высоких температурах происходит также разрыв связей С-С и получаются смеси различных алканов и алкенов, у которых молекулярная масса меньше, чем у исходных алканов (крекинг).

Эти методы имеют промышленное значение.
2. Дегидратация спиртов.
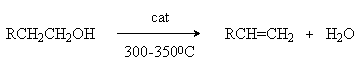
В качестве катализаторов используются Бренстедовские или Льюисовские кислоты, например H3PO4 и Al2O3. В первом случае фосфорную кислоту наносят на пористый носитель, например, силикагель.
Особенностью этих реакций является порядок отщепления воды, определяемый правилом Зайцева: при отщеплении воды наиболее легко отщепляется водород от соседнего наименее гидрированного атома углерода.
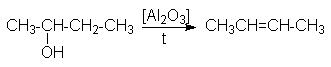
Такой характер отщепления может быть обоснован более высокой термодинамической стабильностью алкенов, у которых наибольшее число заместителей при двойной связи.
Реакции дегидратации имеют препаративное и промышленное значение. Так, в промышленности изобутилен получают дегидротацией изобутилового спирта.
3. Дегидрогалогенирование галогеналканов.
Эта реакция может осуществляться в двух вариантах:
а) щелочное дегидрогалогенирование

Порядок отщепления галогеноводорода в этих реакциях также определяется правилом Зайцева: при отщепление HHal наиболее легко отщепляется водород от наименее гидрированного атома углерода.
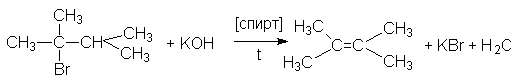
б) термическое дегидрогалогенирование в паровой фазе.
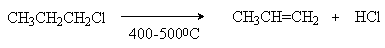
В отличие от щелочного дегидрогалогенирования эта реакция мало избирательна и при наличии нескольких неэквивалентных атомов водорода при вицинальном углеродном атоме образуется смесь алкенов.
Методы имеют препаративное и промышленное значение.
4. Отщепление галогенов от вицинальных дигалогеналканов металлическим цинком.

5. Селективное гидрирование диенов и алкинов.
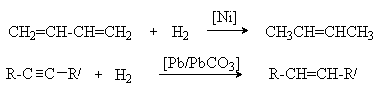
6. Реакция Виттига - взаимодействие карбонильных групп с фосфоний - илидами.
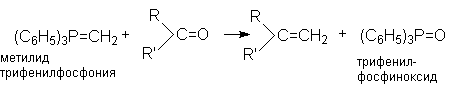
Сами фосфоний - илиды могут быть получены дегидрогалогенированием соответствующим галоидных солей фосфония под действия основания.
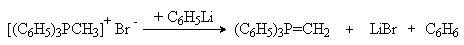
7. Реакция Гофмана - расщепление четвертичных аммониевых солей под действием оснований.
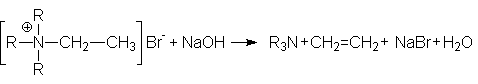
Характер образующихся продуктов определяется правилом Гофмана: при расщеплении образуется наименее замещенный алкен. Например:

Обоснованием правила Гофмана является относительная кислотность атомов водорода при b -углеродных атомах: наименее кислыми являются атомы водорода при наиболее разветвленных углеродных атомах. Так как образующиеся при отрыве от них анионы существенно дестабилизированы алкильными группами, такое отщепление становится энергетически невыгодным.
Физическиесвойства алкенов
Алкены С2-С4- газы, следующие за ними члены гомологического ряда - бесцветные жидкости или кристаллические вещества.
Плотность алкенов выше по сравнению с алканами с тем же числом углеродных атомов. Это обусловлено более сильными межмолекулярными взаимодействиями в алкенах причиной которых являются высокая поляризуемость двойной связи: так RC-C=1,29 a RC=C=4,17.
Высокая поляризуемость двойной связи обусловливает ее значительное поляризацию под действием полярных групп.

Введение алкильных групп у двойной связи увеличивает стабильность молекул алкенов. Это можно проиллюстрировать различием в теплотах гидрирования соответствующих алкенов.
CH2=CH-CH2CH3 + H2 ® CH3CH2CH2CH3 + 127 кДж
транс- CH3-CH=CH-CH3 + H2 ® CH3CH2CH2CH3 + 115 кДж
Такое различие можно проиллюстрировать энергетической диаграммой.

Можно видеть, что транс-псевдобутилену соответствует более низкий уровень энергии и, следовательно, более высокая стабильность.
Электронографическое изучение этилена показало, что геометрия молекулы резко отличается от пространственного строения алканов. Молекула этилена плоская. Углы между связями близки к 120о, связь С=С намного короче связи С-С. Потенциал ионизации в алкенах на 1-1,5 эв меньше, чем у соответствующих алканов, что свидетельствует об электронодонорных свойствах двойной связи.
Подвижность p -электронной системы отражается в электронных спектрах поглощения. Максимум поглощения находится в более длинноволновой области спектра (180-200 нм), чем у алканов. Это связано с переходом электронов со связывающей на разрыхляющую p -орбиталь p ® p *.
В колебательных спектрах кроме поглощения алкильных групп наблюдаются валентные колебания двойной связи при 1640-1660 см-1 и связи =С-Н при 3000-3100 см-1, а также деформационные колебания =С-Н - связи при 890-980 см-1. В спектрах ПМР характерны сигналы при 5,5-6 м.д., соответствующие протонам Н-С=.
Химическиесвойства
Химические свойства алкенов определяются рядом факторов: относительно слабой p -связью в двойной связи, ее поляризуемостью, электронодонорными свойствами, способностью к сопряжению с реакционным центром, возникающим при a -углеродном атоме.
Поэтому естественными реакциями алкенов являются реакции присоединения, среди которых наиболее представительными являются реакции электрофильного присоединения, а также реакции замещения, в которых подвижными являются атомы и группы при a -углеродном атоме по отношению к двойной связи.