|
|
Промисловість реактивів і особливо чистих речовин. 10 страница
Вихід NO при окисненні NH3 на платиновому каталізаторі під атмосферним тиском досягає 98 %, а під тиском до 0,8‑1 МПа і підвищеній температурі – 96 %. Решта NH3 втрачається у вигляді N2.
Рівновага реакції.Для вибору оптимальних умов окиснення NH3 до NO треба насамперед визначити константи рівноваги реакцій (10.4), (10.6) і (10.7) при температурах, близьких до виробничих, а саме 750‑900 °С.
Обчислені за наближеним рівнянням Нернста константи рівноваги цих реакцій мають значення зовсім незначні, тобто всі три реакції необоротні, відбуваються до кінця зліва направо.
Сумарний результат реакцій окиснення NH3, як і будь-якого складного практично необоротного процесу, залежить від співвідношення швидкостей утворення кожного з продуктів при однакових умовах, тому остаточний склад продуктів визначатиметься насамперед селективністю каталізатора.
Каталізатори.Для прискорення реакції окиснення (10.4) використовують платинові каталізатори (платина і сплави платини з платиновими металами) і неплатинові каталізатори (оксиди заліза, марганцю, кобальту та інших металів).
Неплатинові каталізатори дешевші за платинові, але дають менший вихід NO, малоактивні, недовговічні і легко руйнуються кислотами. Найбільш поширеними у промисловості каталізаторами для окиснення NH3 до NO є виготовлені з потрійного (93 % Pt, 3 % Rh, 4 % Pd) або подвійного сплаву (90‑95 % Pt і 5‑10 % Rh) сітки. Каталізатор з потрійного сплаву міцніший і активніший, ніж каталізатор з подвійного сплаву.
Сітки виготовляють з тонкого дроту діаметром від 0,06 до 0,09 мм, з числом плетінь на 1 см2 32×32, що дає 1024 маленьких отвори.
Для окиснення NH3 під атмосферним тиском застосовують пакет з 3‑4 сіток з діаметром від 1,1 до 2,8 м, а під тиском – пакет з 18‑20 сіток діаметром 0,5‑1,15 м.
На платинових каталізаторах NH3 окиснюється дуже швидко, для перебігу реакції потрібні десятитисячні частки секунди. Продуктивність платинового каталізатора і контактного апарату надзвичайно велика. Загальна швидкість процесу окиснення залежно від технологічного режиму і конструкції апарата визначається дифузією NH3 з простору потоку до поверхні каталізатора і тільки при дуже сильному перемішуванні, взаємодією NH3 з О2, адсорбованим на каталізаторі.
Платинові каталізатори надзвичайно чутливі до дії каталізаторних отрут, легко забруднюються домішками, які можуть бути у повітрі або в NH3. Особливо шкідливий для них фосфін, який руйнує платиновий каталізатор незворотно, навіть коли його вміст у газовій суміші близько 0,00001 %. Сірководень руйнує каталізатор тимчасово. Шкідливими є також порох, мастила, часточки оксидів заліза (іржі), які можуть відщеплюватись з поверхні газопроводів, апаратури і летіти разом з газами.
Якщо НNО3 виробляють окисненням NH3 повітрям, усі вихідні продукти треба дуже ретельно очищати від газоподібних і механічних домішок, вміст яких не повинен перевищувати 0,007 мг/м3 газу. Для цього повітря беруть далеко від заводської території, промивають водою, фільтрують крізь сукно, а NH3 крізь шар вати і полотна, і потім змішують. Крім того, газову суміш остаточно очищають перед поданням у контактний апарат, пропускаючи її через керамічні або металокерамічні фільтри, в яких газ очищається більш як на 99 %. З часом на поверхні каталізаторних сіток осідає шар різних часточок, що знижує активність каталізатора. Щоб відновити її, каталізатор промивають розбавленими 15 %-ми розчинами НС1 або НNО3, дистильованою водою і прожарюють у водневому полум'ї.
У процесі роботи каталізатор руйнується. Тонка гладенька нитка його через 10‑20 год. роботи на 10‑20 % потовщується. Це пояснюється тим, що під дією газів поверхня каталізатора стає крихкою, вкривається горбиками і механічно послаблюється. Часточки каталізатора відриваються з поверхні і виносяться з газами. Отже, певна кількість дорогоцінного каталізатора втрачається безповоротно. Частину ж платинового пороху вловлюють на спеціальних фільтрах. Ці втрати платинового каталізатора на установках, що працюють при 750‑850 °С і атмосферному тиску, становлять 0,048‑0,056 г/т 100 %-ної НNО3, а на установках, які працюють під тиском і при температурі близько 900 °С, збільшуються до 0,160‑0,180 г/т НNО3.
Для зменшення втрат платинового каталізатора застосовують двоступеневий каталізатор: І ступінь – сітка з платинового каталізатора; II ступінь – сітка з неплатинового каталізатора. Це дає можливість зберігати приблизно 60 % платинового сплаву і, крім того, знижувати втрати його на 20‑30 % проти втрат при застосуванні тільки платинових каталізаторів.
10.3. Оптимальні умови процесу окиснення амоніаку
Вплив складу вихідної газової суміші.Для створення оптимальних умов контактування найважливішим є співвідношення О2 іNH3 у газовій суміші, яка надходить на контактування. За стехіометричним рівнянням реакції (10.4) на 1 моль NH3 потрібно 1,25 моля О2. Проте, як видно з рис. 10.1, таке співвідношення не забезпечує високого ступеня окиснення NH3. Найбільша швидкість і максимальний вихід NO будуть при співвідношенні NH3 до О2, яке дорівнює 1:1,8‑2, що відповідає вмісту 11‑12 об. % NH3 у газовій суміші. Практично застосовують газову суміш, в якій вміст NH3 дорівнює 10‑11 об. %. Подальше збільшення надлишку О2 помітно не впливає на вихід NO. При застосуванні неплатинових каталізаторів потрібно більше О2. Таке співвідношення NH3 і О2 у газовій суміші зумовлюється ще й тим, що амоніачно-повітряна суміш у певних співвідношеннях стає вибухонебезпечною. На рис. 10.2 показано межі вибухонебезпечного вмісту NH3 у газовій суміші, які з підвищенням температури розширюються. Наявність водяної пари до деякої міри ці межі звужує.

| Рис. 10.1. Залежність виходу NO від співвідношення О2 і NH3 в амоніачно-повітряній суміші | Рис. 10.2. Межі вибухонебезпечного вмісту NH3 в амоніачно-повітряній суміші: 1 – область вибухонебезпечності сухої суміші; 2 – область вибухонебезпечності амоніачно-повітряної суміші над водними розчинами |
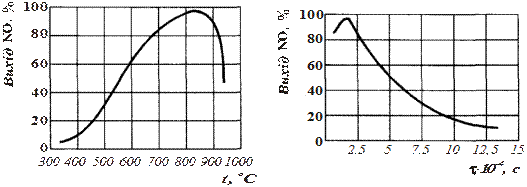
На рис. 10.3 показано залежність виходу NO від температури на платиновому каталізаторі при атмосферному тиску. Як видно, процес окиснення досягає максимального ступеня при температурі 750‑850 °С. При підвищеному тиску оптимальна температура контактування дорівнює 800‑900 °С. Температура каталізаторної сітки завжди приблизно на 75 °С вища, ніж температура газів.
| |
| Рис. 10.3. Залежність виходу NO від температури | Рис. 10.4. Залежність виходу NO від часу контактування |
Залежність виходу NO від часу контактування при атмосферному тиску показано на рис. 10.4. На відміну від багатьох інших процесів, збільшення часу стикання газу з каталізатором (після того, як вихід NO досяг максимальної величини) значно знижує вихід NO. Це пояснюється, можливо, тим, що за більший час відбуваються більш повільні шкідливі побічні реакції. Оптимальний час контактування для високого виходу NO, як це видно з рис. 10.4, змінюється у межах від 0,0001 до 0,0002 с. На неплатинових каталізаторах оптимальний час контактування приблизно у 100 разів більший, ніж на платинових.
На 1 м активної поверхні сітки платинового каталізатора під атмосферним тиском можна окиснювати за добу близько 600 кг, а при підвищенні тиску до 0,8‑1 МПа – до 3000 кг NH3. З цього видно, що продуктивність каталізатора при підвищенні тиску значно зростає. Але з підвищенням тиску збільшуються втрати каталізатора порівняно з установками, що працюють під атмосферним тиском.
При всіх однакових оптимальних умовах вихід NO залежить від кількості каталізаторних сіток. Щоб досягти 98 %-го виходу при атмосферному тиску, треба мати у контактному апараті не менше трьох сіток, а при підвищеному тиску – 18‑20 сіток.
10.4. Переробка нітрозних газів на розбавлену нітратну кислоту
Нітрозні гази, які утворюються внаслідок окиснення NH3 киснем повітря, містять близько 10‑11 об. % NO, 5‑6,5 % О2, N2 і водяну пару. Основний компонент газової суміші – NO – безбарвний газ дуже мало розчиняється у воді і з нею не реагує. Щоб добути HNO3, треба NO окиснити до NO2.
Окиснення NO.Моноксид азоту при звичайних умовах досить легко окиснюється до NO2 за зворотною реакцією (10.1), яка відбувається із зменшенням об'єму і виділенням теплоти. Тому при зниженні температури і підвищенні тиску рівновага зміщується у бік утворення NO2.
При 800‑900 °С окиснення практично не відбувається і навпаки – при 100 °С і нижче NO окиснюється до кінця.
Швидкість і ступінь окиснення NO.Реакція окиснення NO киснем – це реакція третього порядку. Для її здійснення треба, щоб одночасно сполучились три молекули – дві молекули NO і одна молекула О2. Крім того, ця реакція має свою особливість: на відміну від інших хімічних процесів швидкість її при зниженні температури збільшується. Тому для збільшення швидкості окиснення NO треба знижувати температуру нітрозних газів. Взагалі швидкість окиснення NO при будь-яких температурах вимірюється секундами і хвилинами. Ступінь окиснення буде тим більшим, чим нижча температура нітрозних газів. Для пояснення аномального характеру реакції окиснення NO до NO2 запропоновано кілька гіпотез. Одна з них полягає у тому, що окиснення відбувається через утворення проміжного продукту – димера моноксиду азоту
2NO  (NO)2 + Q;
(NO)2 + Q;
(NO)2 +О2 2NO2 + Q.
Утворення димера – процес зворотний, який відбувається з виділенням теплоти. Тому підвищення температури зміщує рівновагу цієї реакції у лівий бік.
Велике значення для процесу окиснення NO має тиск.Азоту (IV) оксид, що утворюється внаслідок реакції окиснення, при низьких температурах полімеризується з утворенням димера
2NO2 N2O4 + 57,0 кДж.
Зниження температури і підвищення тиску зміщує рівновагу реакції у правий бік. Швидкість цієї реакції велика, рівновага встановлюється дуже швидко. При температурах близько –10...–15 °С ступінь полімеризації досягає 98‑99 % і димер перетворюється у рідину. Тому для збільшення швидкості перетворення NO в NO2 реакційну суміш енергійно охолоджують. Підвищення тиску сприяє зміщенню рівноваги реакції вправо і збільшенню швидкості реакції. Тому у виробництві нітратної кислоти в останні роки перейшли від обладнання при атмосферному тиску до обладнання з тиском до 1 МПа.
Абсорбція нітрозних газів водою.Розчинення нітрозних газів у воді супроводиться утворенням нітратної та нітритної кислот:
2NО2 + Н2О = НNО3 + HNO2 + 116,0 кДж; (10.8)
N2O4 + Н2О = НNО3 + HNO2 + 59,0 кДж; (10.9)
N2O3 + Н2О = 2НNО2. (10.10)
Нітритна кислота нестійка і розкладається
3НNО2 = НNО3 + 2NО + Н2О – 75,8 кДж. (10.11)
Підсумовуючи рівняння (10.8) і (10.9), маємо рівняння (10.2). Реакція (10.2) оборотна; стан рівноваги залежить від концентрації НNО3, температури і тиску. Чим менша концентрація НNО3, тим більший ступінь поглинання NO2.
При атмосферному тиску і звичайній температурі утворюється нітратна кислота з концентрацією не більш як 50 % HNO3.
У промисловості ступінь перетворення азоту оксидів в нітратну кислоту при атмосферному тиску досягає 92 %, при тиску 0,6‑0,8 МПа – 98 %. Зменшення температури дає невеликий ефект, бо при зниженні температури зменшується швидкість процесу. Для прискорення взаємодії NO2 з водою збільшують поверхню дотику реагуючих компонентів, заповнюючи башти насадкою.
При підвищеному тиску (0,6‑0,8 МПа) і температурі 50‑80 °С утворюється 58‑60 %-ий розчин HNO3.
Щоб добути розчин НNО3 з концентрацією 98 %, треба збільшити тиск до 5 МПа, а щоб прискорити реакцію, збільшити температуру до 50‑80 °С.
10.5. Виробництво розбавленої нітратної кислоти
Функціональна схема виробництва HNO3 має такі стадії:

У промисловості НNО3 виробляють за трьома технологічними схемами: за першою схемою працюють установки, в яких окиснення NH3 і поглинання оксидів азоту водою відбуваються під атмосферним тиском з виходом 50 %-го розчину НNО3; за другою схемою окиснення NH3 і поглинання оксидів азоту водою відбувається при підвищених тисках (0,6‑0,8 МПа); за третьою схемою NH3 окиснюється при низькому тиску (0,4 МПа), а оксиди азоту поглинаються при підвищених тисках (до 1,2 МПа). Внаслідок цього поліпшується технологія виробництва та отримують НNО3 підвищеної концентрації.
Сучасні технологічні схеми виробництва НNО3, які працюють при підвищеному тиску (від 0,2 до 1 МПа), і комбіновані схеми розроблені за принципом енерго-технологічних систем, в яких енергія відхідних газів і теплота реакції окиснення амоніаку використовується для стискання повітря і нітрозних газів, а також отримання технологічної пари.
Контактні апарати для окиснення NH3 мають різну конструкцію залежно від тиску, під яким відбувається процес окиснення.
Останнім часом почали застосовувати контактні апарати з верхнім введенням газової суміші, схему одного з яких наведено на рис. 10.5. Каталізатори сітки розміщені в апараті на підпірних решітках. Продуктивність такого контактного апарата становить 48‑50 т у перерахунку на HNO3 на добу.

Рис. 10.5. Контактний апарат: 1 – розподільна решітка; 2 – корпус апарата; 3 – платинова сітка; 4 – шар неплатинового каталізатора; 5 – насадка; 6 – опорна решітка
Схему виробництва розбавленої HNO3 при підвищеному тиску (0,73 МПа) подано на рис. 10.6. Повітря, очищене у фільтрі 2, стискується компресором 3 до тиску 0,73 МПа. При цьому газ нагрівається приблизно до 135 °С.
Стиснуте повітря, пройшовши через нагрівач 5, нагрівається до 250 °С завдяки теплоті нітрозних газів і надходить для змішування з NH3 у змішувач 7. Рідкий NH3 зі збірника проходить випарник 6 і подається у змішувач 7. Амоніачно-повітряна суміш вводиться згори у контактний апарат 8, де на Pt-Rh-Pd каталізаторі NH3 окиснюється до NO. При цьому у контактному апараті температура підвищується до 900 °С. Гарячі нітрозні гази, які містять 9‑9,5 % NO, проходять через котел-утилізатор 9, де охолоджуються і відбувається утворення водяної пари.
Далі гази поступають в окиснювач 10, в якому NO окисняється до NO2. Охолоджені у нагрівачі повітря 5, підігрівачі хвостових газів 13 і холодильнику-конденсаторі 12 до температури коло 45 °С нітрозні гази надходять в абсорбційну колону 11, зрошувану водою. Оскільки абсорбція NO2 водою екзотермічна, абсорбційні тарілки мають змієвикові холодильники, в яких циркулює холодна вода. Отримана холодна вода надходить у віддувну колону 12, в якій гарячим повітрям з нітратної кислоти відбуваються розчинені у ній нітрозні гази, які подаються в абсорбційну колону. Хвостові гази, пройшовши систему каталітичного очищення від оксидів азоту відновленням їх амоніаком до N2, викидаються в атмосферу.
В установках такого типу ступінь перетворення амоніаку в нітратну кислоту досягає 98,99 %, а концентрація кислоти – 60,62 %. Однак при окисненні амоніаку під тиском збільшуються втрати платинового каталізатора. Тому застосовують системи, в яких окиснення амоніаку здійснюють при набагато нижчому тиску (~0,4 МПа), ніж окиснення азоту (ІІ) оксиду (до 1,2 МПа).

Рис. 10.6. Принципова технологічна схема отримання розбавленої нітратної кислоти при підвищеному тиску (0,73 МПа): 1 – повітрозабірна труба; 2 – повітроочищувач; 3 – газовий компресор; 4 – газова турбіна; 5 – повітронагрівач; 6 – випаровувач амоніаку; 7 – змішувач з фільтром; 8 – контактний апарат; 9, 17 – котел-утилізатор; 10 – окиснювач з фільтром; 11 – абсорбційна колона; 12 – віддувна колона; 13 – холодильник-конденсатор; 14 – підігрівач "хвостових" газів; 15 – реактор каталітичного очищення; 16 – камера згоряння; 18 – викидна труба
Сучасні азотнокислотні ХТС характеризуються великою потужністю окремої технологічної нитки, що становить 380‑400 тис. т/рік. Вказані системи дозволяють отримати тільки розбавлену нітратну кислоту. Для виробництва вибухових речовин, деякої пластичної маси, барвників потрібна концентрована кислота (98 %). Нітратну кислоту такої концентрації можна отримати або концентруванням розбавленої HNO3, або прямим синтезом.
Відгонкою води з розбавленої HNO3 можна отримати тільки 68 %-ий розчин, оскільки саме така концентрація відповідає азеотропній суміші HNO3‑H2О. Тому концентрування здійснюють із застосуванням водовідбираючих засобів, таких, наприклад, як 92‑94 %-на H2SO4. Концентрована H2SO4, яка утворює при цьому гідрати, кипить при більш високій температурі, ніж 100 %-на НNО3. Як водовідбираючий засіб може використовуватися також магнію азот.
Значне застосування знаходить прямий синтез концентрованої HNO3, який протікає за рівнянням реакції
2N2O4 (р) + 2Н2О (р) + О2 (г) 4HNO3 (Р), DН = – 59,5 кДж.
Насправді поглинання димера азоту диоксиду здійснюється розбавленою нітратною кислотою, що містить коло 45 % води. Ця операція здійснюється в автоклаві при 90 °С і 5 МПа. В автоклаві отримується так званий нітроолеум НNО3·nNО2, що містить до 25 % NO2. Після віддувки азоту диоксиду отримують 97‑98 %-ну нітратну кислоту.
Питання до розділу 10
1. Які способи виробництва нітратної кислоти існували до XX століття?
2. Навести характеристику розбавленої і концентрованої нітратної кислоти.
3. З яких стадій складається отримання нітратної кислоти окисненням амоніаку?
4. Які побічні продукти утворюються при окисненні амоніаку?
5. Які каталізатори використовують при окисненні амоніаку?
6. Яка швидкість каталітичного окиснення амоніаку?
7. Як впливає температура на швидкість окиснення амоніаку?
8. Який оптимальний склад газової суміші при окисненні амоніаку?
9. Які умови окиснення NO до NO2?
10. Які реакції відбуваються при абсорбції нітрозних газів водню?
11. Як впливають температура і тиск на процес утворення HNO3?
12. Які технологічні схеми використовують при виробництві HNO3?
13. Яка конструкція контактного апарата для окиснення амоніаку?
14. Описати схему виробництва розбавленої HNO3 при підвищеному тиску.

Розділ 11. ВИРОБНИЦТВО СУЛЬФАТНОЇ КИСЛОТИ
11.1. Основні властивості та застосування сульфатної кислоти
Безводна H2SO4 (моногідрат) – важка, оливна рідина з густиною 1,8305 г/см3 (при 20 °С), кипить при 279,6 °С і тиску 760 мм рт. ст., кристалізується при 10,37 °С. При нагріванні вище 200 °С безводна H2SO4 частково розкладається за схемою
H2SO4 SO3 + H2O (а)
утворюючи азеотропну суміш, яка містить 98,3 % H2SO4 і 1,7 % Н2О і має температуру кипіння 338,8 °С. Сульфатна кислота змішується з Н2О у будь-яких співвідношеннях, утворюючи гідрати (H2SO4 ∙ Н2О; H2SO4 ∙ 2Н2О; H2SO4 ∙ 4Н2О) з виділенням великої кількості теплоти (937,84 кДж/кг H2SO4). З SО3 сульфатна кислота утворює дві сполуки: H2SO4 ∙ SO3 і H2SO4 ∙ 2SO3. Розчин SO3 в H2SO4 від 18 до 65 % називають олеумом.
Сульфатна кислота надзвичайно активна, вона дуже добре змішується з водою, відбираючи її навіть від хімічних сполук. Рослинні і тваринні тканини, що містять у своєму складі вуглеводи (целюлозу, крохмаль, цукор), обвуглюються концентрованою H2SO4, яка є сильним окисником. У розбавленій H2SO4 целюлоза і крохмаль гідролізує до цукрів. На шкірі людини H2SO4 залишає тяжкі опіки.
Переважна більшість H2SO4 витрачається на виробництво мінеральних добрив. Майже всі інші кислоти і багато солей виробляються за участю H2SO4; її широко використовують у виробництві кольорових і рідкісних металів; у металообробній промисловості H2SO4 та її солі застосовують для протрави металевих виробів перед їх лудінням, нікелюванням, хромуванням або цинкуванням. Значну кількість H2SO4 використовують для очищення нафтопродуктів, у виробництві барвників, лаків, ліків, спиртів, ефірів, синтетичних миючих засобів, отрутохімікатів, вибухових речовин, пластичних мас та ін. Її застосовують у текстильній промисловості для протрави тканин, для сульфування органічних сполук, у промисловості органічного синтезу, для виробництва йонообмінників, штучного волокна. Сульфатну кислоту застосовують у харчовій промисловості для виробництва крохмалю, патоки та інших продуктів, а також для сушіння газів, при концентруванні різних кислот.
Виробництво H2SO4 у світі весь час зростає. Відповідно до державних стандартів виробляється три сорти H2SO4: технічна, акумуляторна і реактивна. Технічна кислота завдяки домішкам має темний колір.
До акумуляторної кислоти ставляться високі вимоги відносно її чистоти. Нормуються вміст Mn, Fe, As, оксидів азоту, важких металів, нелеткого залишку та ін. Акумуляторну кислоту виробляють контактним методом, вона містить 92‑94 % H2SO4.
Реактивну H2SO4 виготовляють трьох марок: хімічно чисту (х. ч.), чисту для аналізу (ч. д. а.) і чисту (ч.). Як правило, її добувають у кварцовій або платиновій апаратурі контактним способом, вона містить 92‑94 % H2SO4.
Відкриття H2SO4 можна віднести до X ст. Щев XIII ст. H2SO4 добували прожарюванням залізного купоросу, завдяки чому і тепер один із сортів H2SO4 називається купоросною оливою. Понад 200 років H2SO4 добували нітрозним способом і більш як 100 років контактним способом.
З кінця XIX ст. набув поширення контактний спосіб виробництва H2SO4 який полягає у тому, що окиснення SO2 до SO3 відбувається на поверхні твердого каталізатора. Контактний спосіб дав можливість добувати олеум і більш чисту H2SO4. Важливим кроком у розвитку контактного способу була заміна дорогого платинового каталізатора ванадієвим, що дало великий економічний ефект і спростило технологію виробництва H2SO4.
Виробництво H2SO4 контактним методом має три стадії:
· очищення газової суміші від домішок-отрут каталізатора;
· окиснення SO2 до SO3 на поверхні твердого каталізатора;
· поглинання SO3 концентрованою H2SO4.
11.2. Сировинна база сульфатно-кислотного виробництва
Незалежно від способів виготовлення H2SO4 першою стадією його є добування сульфітного газу, який утворюється при спалюванні природної S або випалюванні різних сульфітних мінералів: природних сульфітних сполук заліза, міді, цинку, свинцю тощо; гіпсу та ангідриту та ін. Із сульфітних сполук найбільш поширеним є сірчаний колчедан (пірит FeS2), великі поклади якого єна Донбасі. Чистого FeS2 у природі немає. Основними домішками сировини є сульфіди міді, цинку, свинцю, миш'яку, а також селен, телур, тальк, кварц, солі кальцію та магнію, невеликі кількості золота і срібла. Важливим джерелом сировини для добування SO2 є також поліметалічні сульфідні руди кольорових металів – міді, цинку та ін. При добуванні 1 т чорнової міді утворюється 7,3 т SO2, з якого можна добути 11 т H2SO4. Використання газів випаленням руд чорної та кольорової металургії крім економічної доцільності сприяє створенню санітарно-гігієнічних умов праці і запобігає забрудненню навколишнього середовища. Для виробництва H2SO4 використовують також сірчистий газ, який виділяється при переробці кислого гудрону – відходів нафтопереробного виробництва.
Функціональна схема виробництва H2SO4 має такі стадії:

11.3. Виробництво сульфітного газу
У промисловості сульфітним газом називають газову суміш, в якій міститься 7‑15 % SO2, O2 і N2 у різному співвідношенні залежно від якості сировини та умов її випалювання. Сірчистий ангідрид SO2 – це безбарвний газ з характерним різким запахом; подразнює слизові оболонки очей і дихальних шляхів. Він у 2,3 раза важчий за повітря, при температурі –10 °С і атмосферному тиску зріджується, взаємодіючи з водою, утворює нестійку сульфітну кислоту
SO2+H2O H2SO3,
яка існує тільки у водному розчині. Рідкий SO2 застосовують також у холодильній техніці, для вибілювання паперової маси та цукрових розчинів, як консервуючу речовину при транспортуванні та зберіганні плодів та ін. Вміст SO2 у повітрі робочих приміщень не повинен перевищувати 0,1 мг/л.
Сірчаний колчедан випалюють у багатополичкових механічних печах, у печах з киплячим шаром або завислому стані пилеподібного обпалу.
Сумарне рівняння процесу випалювання колчедану з утворенням оксиду заліза таке:
4FeS2 + 11О2 = 2Fe2O3 + 8SO2 + 3415 кДж.
Якщо реакція супроводжується утворенням залізної окалини, то рівняння реакції матиме такий вигляд:
3FeS2 + 8О2 = Fe3O4 + 6SO2 + Q.
Завжди при окисненні FeS2 утворюється плівка оксидів заліза, тому подальше вигорання сірки лімітується дифузією О2 повітря до неокисненого ядра FeS2 і зворотною дифузією SO2 з глибини часточок. Саме цей процес внутрішньої дифузії і визначає загальну швидкість випалення піриту.
Під час випалення колчедану виділяється велика кількість теплоти, тому весь процес відбувається автотермічно. У газову фазу з сировини надходять також As2O3, SeO2 і волога. Твердий залишок після випалення сульфатного колчедану – недопалок (70‑80 % всього колчедану) – містить крім оксидів заліза ще недовипалені FeS2, FeS, негорючі SiO2, CaSO4, MgSO4, іноді золото, срібло, оксиди міді, цинку, кобальту, талію та інших елементів. З недопалку, в якому міститься 0,5‑3 % S і 47‑48 % Fe, виплавляють високоякісний мідистий чавун. Обробляючи недопалок H2SO4 після хлоруючого випалення, з нього вилучають цінні речовини. Крім того, недопалок використовують у цементній промисловості, для виготовлення мінеральних фарб (сурик, мумія), як шліфувальний порошок при виготовленні дзеркального скла.
Рушійна сила процесу випалення зростає із збільшенням вмісту FeS2 у природній руді і концентрації О2 у газовій суміші. Як правило, використовують майже подвійну кількість повітря. Збільшення швидкості реакції завдяки зростанню коефіцієнта масопередачі К досягається підвищенням температури. Але таке підвищення обмежується спіканням часточок колчедану, яке настає при 850‑1000 °С залежно від домішок до колчедану і виду випалювальної печі. Дисоціація колчедану починається при температурі понад 500 °С і з підвищенням її швидко зростає. Швидкість реакції окиснення сульфітних сполук обернено пропорційна розмірам кусків колчедану і зростає з підвищенням температури. Дифузійні процеси інтенсифікуються перемішуванням колчедану у повітрі. Проте процес вигорання колчедану лімітується в основному дифузією О2 і SO2 у порах оксиду заліза. Тому для полегшення дифузії і збільшення поверхні контактування FeS2 з О2 повітря краще подрібнювати колчедан. Флотаційний колчедан складається в основному з часточок розміром від 0,03 до 0,3 мм. Розмір грудочок колчедану при спалюванні у поличкових печах становить 3‑4 мм. Із зменшенням розміру часточки колчедану починають спікатись у поличкових печах при нижчій температурі. Під час випалювання колчедану у завислому стані та у киплячому шарі процес спікання спостерігається набагато рідше. Отже, швидкість випалювання колчедану зумовлюється швидкістю дифузії О2, який у шарі реагуючих компонентів дифундує до поверхні часточок через суміш SO2 і N2 повітря, що рухається від поверхні у газову фазу.