|
|
Химические свойства нитросоединений.
Химические свойства нитросоединений определяются природой NO2-группы и ее влиянием на R. Наиболее характерная реакция NO2-группы – ее восстановление. Основность NO2-группы низка, с донорами протонов она образует слабые водородные связи. Свойства нитросоединений рассматривают с учетом того, с каким атомом С связана NO2-группа. В насыщенных соединениях (СSP3-NO2) NO2-группа активирует α-водородные атомы (С-Н-кислотность), что приводит к реакциям по α-углеродному атому. В молекулах сопряженных нитроалкенов (СSP2-NO2) двойная связь С=С оказывается сильно поляризованной, что облегчает реакции нуклеофильного присоединения и полимеризации. В аренах NO2-группа дезактивирует кольцо, затрудняя электрофильное и облегчая нуклеофильное замещение в бензольном ядре.
Основные реакции нитросоединений:
6.1. восстановление NO2 –группы;
6.2. реакции по α-углеродному атому;
6.3. реакции по ароматическому кольцу: реакции электрофильного замещения SE и реакции нуклеофильного замещенияSN.
6.1. Восстановление NO2 –группы.
В зависимости от условий реакции и применяемых восстановителей можно получить различные продукты нитросоединений.

Процесс восстановления ступенчатый, протекает через образование нитрозосоединений, гидроксиламинов и далее до аминов:

Если в α-положении к нитрогруппе есть атомы Н, то имеет место нитрозо-оксимная таутомерия:
 .
.
Нитрозалканы восстанавливаются далее через алкилгидроксиламины в амины:

Все нитросоединения можно восстановить до аминов. Для осуществления превращения нитроалканы → алкиламины в качестве восстановителей используют: Fe или Sn в кислой среде, NH2NH2 или гидриды металлов (LiAlH4).
 .
.
Для превращения нитроарены→ариламиныиспользуют следующие реагенты: H2 (Cu+/Al2O3), металлы и их соли в присутствии кислот (Zn, Sn, Fe), Na2S, (NH4)2S, Na2S2O3, растворы Na в спиртах, растворы Zn или SnCl2 в щелочах, гидриды металлов (LiAlH4, NaBH4), NH2NH2 в присутствии катализаторов или без них. Например:

Наиболее изученной является реакция восстановления именно нитробензола. Впервые Н.Н.Зинин в 1842 г. осуществил восстановление нитробензола с использованием Н2S/NH3 или (NH4)2S.

Важно отметить, что поскольку описанные выше превращения относятся к окислительно-восстановительным процессам, глубина протекания восстановления существенным образом зависит от природы заместителя в бензольном кольце, при этом донорные заместители снижают способность нитросоединений к восстановлению, а акцепторные, наоборот, повышают. Это означает, что при восстановлении замещенных аренов конкретными восстановителями, восстанавливающими нитробензол до анилина, соответствующие замещенные амины могут быть получены, если электронные свойства арильной группы близки к фенильной. В противном случае можно ожидать образования других продуктов. В качестве примера можно привести реакцию восстановления 1,3-динитробензола гидросульфидом натрия.

Независимо от избытка восстановителя реакция завершается восстановлением только одной нитрогруппы, поскольку в ходе реакции происходит переход от активирующей восстановление нитрогруппы как заместителя к дезактивирующей аминогруппе после восстановления первой нитрогруппы, что полностью исключает дальнейшее восстановление второй нитрогруппы. С этих позиций становится понятным региоселективность восстановления 2,4-динитрофенола сульфидом аммония: индуктивный эффект гидроксильной группы способствует повышению реакционной способности нитрогруппы в орто-положении в отношении восстановителя:

Следует отметить, что глубина протекания процесса восстановления металлами зависит от кислотности среды – в более кислой среде при прочих равных условиях восстановление протекает более глубоко. Активность восстанавливающих агентов меньше в нейтральной и щелочной, чем в кислой среде и реакцию можно остановить на разной степени восстановления нитро-группы. Последовательность продуктов, образующихся промежуточно при восстановлении нитробензола железными стружками в нейтральной или слабокислой среде, представлена ниже:

Препаративное значение имеет синтез фенилгидроксиламина восстановлением нитробензола, который осуществляют в слабокислой среде, используя в качестве восстановителя цинк в растворе хлорида аммония:
 .
.
Нитрозобензол из нитробензола удается получить при действии цинка в водной среде или электрохимическим восстановлением:
 .
.
Своеобразными восстановителями нитросоединений являются эфиры фосфористой кислоты – триалкоксифосфиты. Их действие на нитросоединения сводится к тому, что эти восстановители отщепляют по одному атому кислорода от нитрогруппы, приводя последовательно к нитрозосоединениям и нитренам, которые трансформируются далее с учетом структуры восстанавливаемого соединения:

Помимо продуктов прямого восстановления нитросоединений могут образовываться и продукты взаимодействия промежуточных соединений между собой. Так, промежуточно образующееся нитрозосоединение может реагировать с конечным продуктом – амином с образованием азосоединения.

Реакция нитрозосоединения с другим интермедиатом –арилгидроксиламином – приводит к азоксисоединению.

Восстановление азосоединения может происходить в условиях реакции с образованием гидразосоединения. Однако, выбор условий реакции (восстановителя, рН среды) позволяет получать не весь набор возможных продуктов восстановления одновременно, а почти исключительно только один из них. Разработаны препартивные способы получения перечисленных выше соединений из нитробензола: 
При взаимодействии ароматических нитросоединений с оксидом мышьяка (III) в щелочной среде или с метилатом натрия в метанольном растворе образуются азоксисоединения. Восстановление цинком в щелочной среде в растворе этилового спирта приводит к азосоединениям, которые могут быть окислены в азоксисоединения действием пероксида водорода и, наоборот, азоксисоединения восстанавливаются в азосоединения трифенилфосфином. Восстановление цинком в водно-спиртовом растворе гидроксида натрия приводит к гидразосоединениям, которые под действием кислорода воздуха легко окисляются в азосоединения.
6.2. Реакции по α-углеродному атому.
6.2.1. С-Н кислотность первичных и вторичных нитроалканов и их таутомерные превращения
Электроноакцепторные свойства NO2 –группы обусловливают повышенную кислотность С-Н-связей в α-положении. Значения С-Н-кислотности ряда нитроалканов
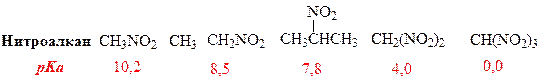
Указанные значения рКа относятся только к отщеплению протона, принадлежащего α-углеродному атому. Вследствие этого нитросоединения, содержащие водород в α-положении, растворяются в водной щелочи, хотя в нейтральной водной среде они не растворимы (кроме низших гомологов). Таким образом, первичные и вторичные нитросоединения можно отличить от третичных, поскольку последние не растворяются в водных растворах щелочей.
Только в случае отщепления протона от α-углеродного атома образуется сопряженное основание, которое может быть стабилизировано с участием NO2-группы.

Образовавшийся анион называется анионом нитрония– амбидентный ион. Протонирование аниона нитросоединения может происходить по двум центрам – α-атому углерода и атому кислорода нитрогруппы. Поскольку кинетически протонирование происходит по более отрицательному атому кислорода, то при протонировании аниона образуется не само нитросоединение, а его таутомер, называемый ациформой или нитроновой кислотой.

Нитро-форма и аци-нитроформа являются изомерами, находящимися в состоянии динамического равновесия. Это явление называется таутомерией, а сами изомеры – таутомерами.
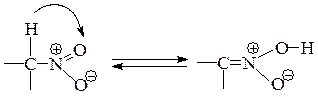
Отметим, что анион нитросоединения и его ациформа обладают электронным строением аналогичными карбоксилат-аниону и карбоксильной группе.
В общем случае, нитроформа для подавляющего большинства нитросоединений является более устойчивой, чем ациформа, однако в растворе нитросоединение часто находится в таутомерном равновесии со своей ациформой, причем относительная концентрация последней заметно выше, чем концентрация енольной формы в растворе карбонильного соединения. Иногда нитросоединение удается выделить в виде метастабильной ациформы. Так, при аккуратном подкислении щелочного водного раствора фенилнитрометана выпадает кристаллический осадок ациформы последнего.

При стоянии это кристаллическое вещество становится жидкостью вследствие его превращения в смесь нитроформы, которая преобладает, и ациформы.
Представляет интерес обратимое фотохимическое превращение орто-нитротолуола в его ациформу, имеющую хиноидную структуру. При этом происходит изменение цвета желтого нитросоединения на синий цвет аци-нитро формы.

При отсутствии облучения в темноте происходит обратное превращение. Это позволяет использовать подобные вещества в качестве фотохромных красителей, например при производстве фотохромных стекол для солнцезащитных очков.
6.2.2. Превращения нитроалканов в присутствии сильных кислот.
В сильнокислой среде (80-95% H2SO4) при нагревании нитросоединения перегруппировываются в гидроксамовые кислоты. Полагают, что реакция происходит через ациформу. Гидроксамовые кислоты – производные карбоновых кислот гидролизуются в кислой среде с образованием карбоновых кислот и соли гидроксиламина:
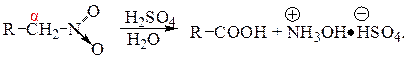
Механизм реакции включает внутримолекулярный окислительно-восстановительный перенос кислорода от N к C и образование гидроксамовой кислоты:

Один из промышленных методов получения гидроксиламина базируется на этой реакции с использованием динитроэтана-1,2 в качестве исходного соединения.
Вторичные нитроалканы в этих условиях реагируют с образованием оксимов:
 .
.
Под действием сильных кислот нитроновые кислоты гидролизуются с образованием альдегидов или кетонов.

Эта реакция известна под названием реакция Нефа.
6.2.3. Галогенирование в щелочной среде.
Галогенирование нитросоединений протекает с замещением водорода при α-углеродном атоме в щелочной среде.
При бромировании аниона нитросоединения могут быть выделены только продукты термодинамического контроля, образующиеся в результате электрофильной реакции по атому углерода.

Так, при взаимодействии нитропроизводных с бромом в щелочной среде в зависимости от соотношения реагентов могут быть получены моно- или дибромпроизводные.
6.2.4. Реакция с азотистой кислотой.
Первичные и вторичные нитросоединения можно различить между собой реакцией с азотистой кислотой. Для этого нитросоединение растворяют в водном растворе щелочи, добавляют нитрит натрия и осторожно подкисляют полученный раствор. В случае первичных нитросоединений образуется осадок так называемой нитроловой кислоты – геминального нитрооксима. Этот осадок растворим в водной щелочи с образованием раствора красного цвета, который обусловлен присутствием аниона нитроловой кислоты.

Вторичные нитросоединения также реагируют с азотистой кислотой, при этом образующийся псевдонитрол (вещество сине-зеленого цвета) не обладает кислыми протонами и в водных растворах щелочей не растворяется.

Третичные нитросоединения с азотистой кислотой не реагируют.
6.2.5. Конденсация нитроалканов с альдегидами и кетонами.
Поскольку при наличии протонов у α-углеродного атома нитросоединения легко образуют анионы в щелочной среде, они могут выступать в качестве активных метиленовых компонент в реакциях конденсации. Так, нитросоединения легко реагируют в щелочной среде с карбонильными соединениями. Реакция происходит через промежуточное образование продукта конденсации альдольного типа, который в условиях реакции подвергается дегидратации, давая продукт кротонового типа – нитроолефин (реакция Генри).

Все стадии реакции являются обратимыми.
6.2.6. Нитроалканы в реакции Михаэля.
Анионы, генерированные из нитроалканов, способны выступать в качестве нуклеофилов в реакции Михаэля, присоединяясь по активированным связям С = С.

6.3. Реакции нитроалкенов по кратной связи.
Для сопряженных нитроалкенов характерны реакции по С=С связи:
6.3.1.Гидрирование.
Восстановление кратной связи можно осуществить селективно, не затрагивая нитрогруппы:

Гидрирование на никеле Ренея приводит к образованию алкиламина.
6.3.2. Присоединение нуклеофилов (спирты, амины).
Поляризация кратой связи, вызванная π-π сопряжением с соседней нитрогруппой, облегчает нуклеофильное присоединение спиртов, аминов, воды.
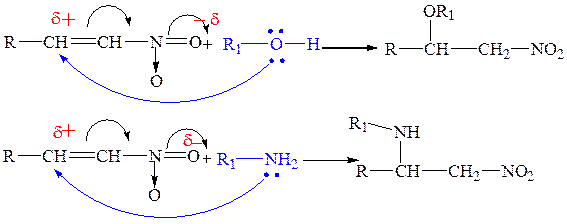
Реакции такого типа называют нитроэтилированием.
Присоединение воды сопровождается расщеплением промежуточного нитроспирта. Это превращение представляет ретро-реакцию конденсации нитросоединений с карбонильными соединениями, которая рассмотрена выше.

6.4. Реакции нитроаренов по ароматическому кольцу (SЕиSN-реакции).
6.4.1.Реакции электрофильного замещения (SЕ-реакции).
Нитрогруппа, являясь сильным акцептором электронов как по индуктивному, так и по мезомерному эффектам, сильно дезактивирует ароматическое кольцо по отношению к реакциям ароматического электрофильного замещения

Так, нитробензол не вступает в реакции алкилирования и ацилирования по Фриделю-Крафтсу, и поэтому может использоваться как растворитель для проведения этих реакций. Нитроарены не сочетаются с солями арилдиазония, не нитрозируются азотистой кислотой.
Нитроарены в SE-реакциях взаимодействуют только с сильными электрофильными реагентами и при повышенных температурах (реакции нитрования,сульфирования, бромирования, хлорирования). Бромирование и хлорирование осуществляют в присутствии кислот Льюиса; нитрование проводят в жестких условиях с применением в качестве нитрующих агентов смеси «дымящей» азотной кислоты и олеума или нитратов с концентрированной серной кислотой; сульфирование осуществляют олеумом при высокой температуре. Во всех этих реакциях электрофильный реагент замещает водород в нитробензоле в мета-положении.

6.4.2. Реакции нуклеофильного замещения (SN-реакции).
Нитрогруппа, расположенная в бензольном кольце, особенно в орто- и пара-положениях к уходящей группе, облегчает протекание ароматического нуклеофильного замещения.

Так, замещение хлора в хлорбензоле под действием водной щелочи с образованием фенола идет при температуре около 300 °С, в то время как в 2,4,6-тринитрохлорбензоле это превращение происходит при комнатной температуре, приводя к пикриновой кислоте.

Легкость протекания реакции нуклеофильного замещения объясняется стабилизацией анионного σ-комплекса (комплекса Мейзенгеймера) нитрогруппой.

В некоторых случаях высоко стабилизированные анионы можно выделить – комплексы Мензенгеймера.

Стабилизация аниона нитрогруппой возможна при ее расположении в о- или п-положениях по отношению к атакуемому нуклеофилом атому углерода в бензольном ядре. В этой связи становится понятным результат реакции:
 .
.
Кроме галогена, в SNреакциях нитрозамещенных аренов уходящими группами могут быть сульфо-, нитро-, алкокси, амино-, аммоннийная группы и даже гидрид-ион. К примеру, при взаимодействии полинитробензолов с О-нуклеофилами нитрогруппа способна выступать и в качестве уходящей группы.

Нитрогруппу можно аналогично заместить на -OR ,-ОAr,-NH2 группы.
Незамещенные мононитроарены реагируют с сильными нуклеофилами только в жестких условиях:
 .
.
Особенно легко протекает замещение галогена в молекулах, содержащих несколько NO2-групп. На этом основана реакция определения концевых аминокислот в пептидах по Сэнгеру:
