|
|
Химические свойства солей арилдиазония.
Соли арилдиазония обладают высокой реакционной способностью. Формально реакции ароматических диазосоединений можно разделить на две большие группы:
· реакции, идущие с выделением азота;
· реакции, идущие с сохранением азота.
5.1. Реакции солей диазония, идущие с выделением азота.
Это самая разнообразная и важная в синтетическом плане группа реакций солей диазония. Их протекание объясняется высокой устойчивостью образующейся молекулы азота.
Реакции солей диазония с выделением азота могут протекать как по гетеролитическому, так и по гомолитическому механизмам. Характер разрыва связи С–N в катионе арилдиазония зависит главным образом от природы нуклеофильного агента. Гетеролитический разрыв этой связи имеет место при взаимодействии с жесткими нуклеофильными реагентами – водой, фторид-ионом. Гомолитическому расщеплению связи С–N благоприятствует использование мягких нуклеофильных агентов (иодид-ион, циано-группа) и рост нуклеофильных свойств растворителя. Для многих реакций переход от гетеролитического к гомолитическому механизму происходит легко при введении в раствор солей одновалентной меди, а иногда даже при введении электроноакцепторного заместителя в бензольное кольцо катиона диазония.
5.1.1 Синтез фенолов из солей диазония. Универсальным методом замены аминогруппы на гидроксил в ароматическом ряду является диазотирование первичного амина с последующим разложением соли диазония. Замещение диазогруппы на НО-группу проводят в водном 40-50%-м растворе серной кислоты, к которому при кипении постепенно добавляют раствор соли диазония. Выходы фенолов в зависимости от природы заместителей в бензольном кольце изменяются от 70 до 90%.
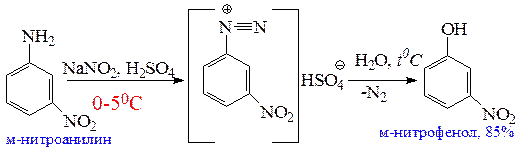
Реакция протекает по мономолекулярному механизму. Образование фенола из соли диазония является единственным примером реакции нуклеофильного ароматического замещения, в которой в качестве интермедиата образуется арилкатион.
Механизм реакции - SN1Ar:

Серную кислоту берут вместо HCl или HBr при замене диазогруппы на гидроксигруппу, так как возможно конкурентное взаимодействие с другими нуклеофилами, а образующийся в сернокислотной среде эфир (из Ar+ и HSO4–) в условиях реакции полностью гидролизуется до фенола.
5.1.2 Реакция Шимана. Замещение диазогруппы на фтор происходит при термическом разложении сухого тетрафторбората арилдиазония:

Использование сухих солей объясняется возможностью при наличии следов влаги протекания побочной реакции образования фенола, который при высоких температурах подвергается осмолению. Реакция идет по ионному механизму через промежуточный арилкатион.

Механизм реакции замещения диазогруппы на фтор напоминает механизм замещения диазогруппы на НО-группу. Выходы арилфторидов сильно зависят от природы заместителя в бензольном кольце арилдиазония. При наличии сильных электроноакцепторных заместителей выход арилфторидов оказывается низким, а если в кольце содержится электронодонорный заместитель выходы оказываются удовлетворительными.
Пример использования реакции: 
 .
.
5.3 3. Замена диазогруппы на иод. Замещение диазогруппы на иод относится к наиболее простым реакциям ароматических диазосоединений, поскольку не требует присутствия катализатора.

Предполагается, что реакция идет по ион-радикальному механизму. Происходит перенос одного электрона от аниона иода к катиону диазония:

Зарождение цепи начинается в результате взаимодействия катиона диазония с иодид-ионом с образованием диазорадикала и атомарного иода. Диазорадикал распадается с выделением азота, а образующийся арильный радикал рекомбинирует с иодом.
5.1.4. Реакция Зандмейера. Для получения арилхлоридов, бромидов, цианидов из солей диазония используют реакцию Зандмейера – взаимодействие соли арилдиазония с хлорид-, бромид- или цианид-ионом, соответственно, в присутствии ионов меди (I).
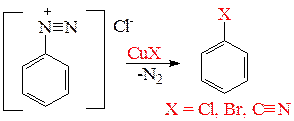
Для этой реакции предполагается радикальный механизм, при котором ион меди (I) играет роль переносчика электронов.

Примеры использования:
а) замена диазогруппы на бром
 ;
;
б) замена диазогруппы на циано-группу

5.1.5. Реакция Гаттермана. Замена диазогруппы на нитрогруппу в тетрафторборатах арилдиазония, катализируемая порошком меди, получила название реакции Гаттермана:

5.1.6. Реакция Гомберга-Бахмана. Разложение солей арилдиазония, содержащих электроноакцепторные группы, в присутствии аммиакатов меди (I) приводит к образованию симметричных биарилов (реакция Гомберга-Бахмана):
 .
.
Для синтеза несимметричных биарилов используют разложение солей арилдиазония в слабощелочной среде в присутствии избытка ароматического соединения (однако выход несимметричных биарилов невысок (до 30%):
 .
.
При арилировании замещенного бензола всегда образуется смесь продуктов, в которой преобладают о- и п-замещенные бифенилы независимо от природы заместителя в бензольном кольце.
5.1.7. Арилирование алкенов по Меервейну. Реакция Меервейна – введение арильной группы в непредельные соединения при взаимодействии их с солями арилдиазония. Реакция может идти путем замещения атома водорода при ненасыщенном атоме углерода на арилрадикал (арилирование) или путем присоединения к кратной связи арилрадикала и галогена (так называемое галогенарилирование).

Соединения с активированной электроноакцепторными группами кратной связью более реакционноспособны. Реакция идет по радикальному механизму через образование арилрадикала в качестве интермедиата.
5.1.8. Дезаминирование. Одним из методов дезаминирования, то есть замены аминогруппы на водород, является диазотирование амина и последующее взаимодействие соли диазония со спиртом как донором водорода:

Реакция протекает по цепному радикальному механизму.
При разложении солей арилдиазония в спиртах помимо арена возможно образование арилалкилового эфира. Замещению диазогруппы на водород способствуют электроноакцепторные заместители в ароматическом ядре; электронодонорные облегчают образование эфиров. В ряде случаев соотношение продуктов можно контролировать варьированием рН раствора:
Универсальным восстановителем при замещении диазогруппы на водород является фосфорноватистая кислота H3PO2:
 .
.
Для достижения хорошего выхода арена обычно используют 5-кратный избыток кислоты. Замещение диазогруппы протекает по радикальному механизму, который можно представить схемой:

Пример использования:

Кроме фосфорноватистой кислоты для замещения диазогруппы на водород можно использовать также NaBH4, при этом другие группы, (CHO, галогены и пр.) не затрагиваются.
5.2. Реакции солей диазония без выделения азота.
Соли арилдиазония вступают в разнообразные реакции, в результате которых атомы азота или даже диазогруппа остаются в продукте реакции.
5.2.1. Восстановление солей диазония. Восстановление солей арилдиазония приводит к арилгидразинам. В качестве восстановителя используется смесь сульфита и гидросульфита натрия

Механизм реакции:

Если в катионе диазония отсутствуют другие группы, чувствительные к восстановлению, то в качестве восстановителя можно использовать SnCl2 в соляной кислоте.
5.2.2. Окисление солей арилдиазония. При окислении солей арилдиазония мягкими окислителями (H2O2, K3Fe(CN)6, NaOCl), образуются N-нитроариламиносоединения, которые под влиянием кислот перегруппировываются в о- и п-нитроанилины: 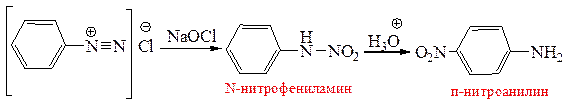
5.2.3. Реакция азосочетания. Из реакций солей диазония, протекающих без выделения азота, важнейшей является азосочетание.
Азосочетание – взаимодействие солей арилдиазония с веществами, содержащими способный замещаться атом водорода, связанный с углеродом, которое приводит к азосоединениям.
Эта реакция имеет большое значение в химии красителей, так как образующиеся азосоединения имеют интенсивную окраску.

Соль диазония в реакции азосочетания называют диазосоставляющей(диазокомпонентой), а ароматический субстрат – азосоставляющей(азокомпонентой).
Азосочетание представляет реакцию электрофильного замещения и протекает по механизму SE:

Диазониевые соли выступают в качестве электрофильного агента, однако катион диазония является слабым электрофилом, поскольку положительный мезомерный эффект фенильной группы уменьшает положительный заряд на атоме азота. Однако активность диазосоставляющей можно повысить введением электроноакцепторных групп в орто- или пара-положения. Так, введение в пара-положение нитрогруппы увеличивает скорость реакции азосочетания в 1300 раз по сравнению с незамещенным реагентом.
В качестве азосоставляющей могут выступать только активированные ароматические соединения, например, фенолы, нафтолы, ароматичекие амины, реакционноспособные гетероциклы (фуран, тиофен и др.).
При планировании реакции азосочетания необходимо учитывать влияние на возможность протекания реакции не только электронных эффектов, но и пространственных факторов. Так, 2,6,N,N-тетраметиланилин несмотря на присутствие сильного электроно-донорного заместителя диметиламиногруппы в реакцию азосочетания не вступает. Это объясняется тем, что диметиламиногруппа выведена из плоскости сопряжения с бензольным кольцом и не оказывает активирующего влияния (отсутствует +М эффект).

Скорость реакции азосочетания (особенно с фенолами) существенно зависит от рН среды. Поведение фенолов и аминов в реакции азосочетания объясняется тем, что диазониевый ион вследствие слабой электрофильности способен эффективно атаковать лишь свободный амин и фенолят-анион. При низких значениях рН ароматический амин находится преимущественно в виде аммониевой соли, а атом кислорода в феноле протонирован. При этом соль диазония, будучи слабым электрофилом, практически не атакует азосоставляющую, так как положительный заряд на атомах азота или кислорода в кислой среде резко понижает нуклеофильность субстрата. По мере возрастания рН увеличивается концентрация свободного амина и фенола и растет скорость реакции. При значениях рН больше 7 концентрация свободного амина достигает максимума, а в случае фенолов происходит образование фенолят-аниона, который является более сильным нуклеофилом, чем фенол. Резкое снижение скорости реакции при рН более 10 связано с переходом реагента в неактивную форму из-за образования ковалентных диазогидроксидов и диазотатов, не обладающих электрофильностью.
Азосочетание с фенолами проводят, как правило, в нейтральной
или слабощелочной среде при рН=7-9, когда концентрация фенолят-аниона достаточно высока.

Сочетание с аминами проводят в слабокислых растворах, когда концентрации катиона диазония и свободного амина достаточно высокие.

Азосочетание аминофенолов с несогласованной ориентацией заместителей можно проводить селективно, меняя рН среды:

Реакция азосочетания является достаточно селективной. В ходе изучения реакции азосочетания были сформулированы эмпирические правила, позволяющие предсказать строение преимущественно образующегося продукта. Эти правила известны как «правила азосочетания»:
– диазонийкатион атакует пара-положение азокомпоненты. Если пара-положение занято группами -COOH, -SO3H, то они отщепляются, если же в пара-положении имеются другие заместители, то реакция идет в орто-положение;
– α-нафтолы сочетаются в положение 4; если оно занято, то в положение 2;
– β-нафтолы сочетаются только в положение 1. Если оно занято группами COOH, -SO3H, NHCH3, Hal, то они замещаются азогруппой, а если СН3 группой, то реакция азосочетания не идет;
– орто- и пара-нитрофенолы, нитроанилины, мета-хлоранилин, мета-аминобензолсульфокислота как азосоставляющие обычно не вступают в реакцию;
– гидрохинон, пирокатехин, пара-фенилендиамин, орто- и пара-аминофенолы окисляются солями диазония до хинонов.
Примеры синтезов.
Получение метилового оранжевого (гелиантина):

Укажите, какую азо- и диазо-компоненты следует использовать для получения метилового красного реакцией азосочетания?

Рассмотрим реакцию получения метилового красного. Очевидно следует проанализировать его структуру для выбора соединений, которые будут использованы в качестве исходных веществ для получения диазо- и азосоставляющих компонентов реакции азосочетания.
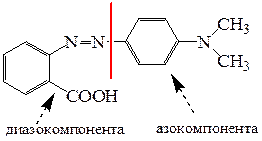
Схема синтеза:

5.2.4. Образование триазенов. В средах, близких к нейтральной, при взаимодействии с первичными или вторичными аминами становится возможной атака катиона диазония как по атому углерода, так и по атому азота. При взаимодействии по атому азота образуются диазоаминосоединения – триазены:

В кислой среде триазены перегруппировываются в 4-(ариалазо)-анилины:

Несимметричные триазены существуют в виде двух таутомерных форм с преобладанием той, в которой протон находится у более основного атома азота:

Под действием концентрированных кислот триазены разлагаются с образованием более основного из двух возможных амина, а «азофрагмент» превращается в соль диазония:
