|
|
Замещение галогена в ароматических соединениях.

Замещение галогена на гидроксильную группу протекает в жестких условиях и известно как «Дау»-процесс (1928 г.)
Раньше этим способом получали фенол (из хлорбензола), но теперь его значение снизилось в связи с разработкой более экономичных способов, не связанных с затратами хлора и щелочи и образованием большого количества сточных вод.
В активированных галогенаренах (содержащих наряду с галогеном нитрогруппу в о– и п-положениях) замещение галогена протекает в более мягких условиях:

Это можно объяснить электроноакцепторным влиянием нитрогруппы, которая оттягивает на себя электронную плотность бензольного кольца и таким образом участвует в стабилизации σ-комплекса:

3. Способ Рашига. Это видоизмененный хлорный метод: бензол подвергается окислительному хлорированию действием хлористого водорода и воздуха, и затем, не выделяя образовавшийся хлорбензол, гидролизуют его водяным паром в присутствии солей меди. В результате хлор вообще не расходуется, а суммарный процесс сводится к окислению бензола в фенол:
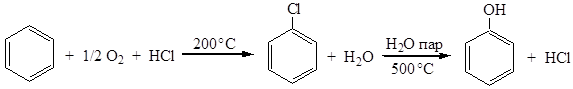
4. Сульфонатный способ. Фенолы можно получить с хорошим выходом при сплавлении ароматических сульфокислот Ar-SO3H со смесью гидроксидов натрия и калия (реакция щелочного плавления) при 300°С с последующей нейтрализацией образующегося алкоголята путем добавления кислоты:

Метод эксплуатируется в промышленности до сих пор (для получения фенола) и используется в лабораторной практике.
5. Кумольный метод. Первое крупное производство фенола кумольным методом было осуществлено в 1949 г. в Советском Союзе. Метод был разработан Р.Ю. Удрисом, Б.Д. Круталовым и др.; в настоящее время это основной метод получения фенола и ацетона.
Метод включает две стадии: окисление изопропилбензола (кумола) кислородом воздуха до гидропероксида и его кислотное разложение:

Преимуществом данного метода является отсутствие побочных продуктов и высокая потребность в конечных продуктах – феноле и ацетоне.
6. Из солей диазония. Метод заключается в нагревании солей диазония в разбавленной серной кислоте, что приводит к гидролизу – замене диазогруппы на гидроксигруппу. Синтез весьма удобен для получения гидроксиаренов в лабораторных условиях:

Строение фенолов
Строение и распределение электронной плотности в молекуле фенола можно изобразить следующей схемой:

Дипольный момент фенола составляет 1.55 Д и направлен в сторону бензольного кольца. Гидроксильная группа по отношению к бензольному кольцу проявляет –I эффект и +М эффект. Так как мезомерный эффект гидроксигруппы преобладает над индукционным, сопряжение неподеленных электронных пар атома кислорода с p-орбиталями бензольного кольца оказывает электронодонорное влияние на ароматическую систему, что повышает ее реакционную способность в реакциях электрофильного замещения.
Химические свойства фенолов
Химические свойства фенолов определяются наличием в молекуле гидроксильной группы и бензольного кольца.
1. Реакции по гидроксильной группе. Фенолы, так же как и алифатические спирты, обладают кислыми свойствами, т.е. способны образовывать соли – феноляты. Однако они более сильные кислоты и поэтому могут взаимодействовать не только со щелочными металлами (натрий, литий, калий), но и со щелочами и карбонатами:

Константа кислотности рКа фенола равна 10. Высокая кислотность фенола связана с акцепторным свойством бензольного кольца (эффект сопряжения) и объясняется резонансной стабилизацией образующегося фенолят-аниона. Отрицательный заряд на атоме кислорода фенолят-аниона за счет эффекта сопряжения может перераспределяться по ароматическому кольцу, этот процесс можно описать набором резонансных структур:

Ни одна из этих структур в отдельности не описывает реального состояния молекулы, но их использование позволяет объяснять многие реакции.
Феноляты легко взаимодействуют с галогеналканами и галогенангидридами:

Взаимодействие солей фенола с галогеналканами – реакция О-алкилирования фенолов. Это способ получения простых эфиров (реакция Вильямсона, 1852 г.).
Фенол способен взаимодействовать с галогенангидридами и ангидридами кислот с получением сложных эфиров (О-ацилирование):

Реакция протекает в присутствии небольших количеств минеральной кислоты или при нагревании.
2. Реакции по бензольному кольцу. Гидроксил является электронодонорной группой и активирует орто– и пара-положения в реакциях электрофильного замещения:

Галогенирование
Галогенирование фенолов действием галогенов или галогенирующих агентов протекает с большой скоростью:


Нитрование
При действии азотной кислоты в уксусной кислоте (в присутствии небольшого количества серной кислоты) на фенол получается
2-нитрофенол:

Под действием концентрированной азотной кислоты или нитрующей смеси фенол интенсивно окисляется, что приводит к глубокой деструкции его молекулы. При использовании разбавленной азотной кислоты нитрование сопровождается сильным осмолением несмотря на охлаждение до 0°С и приводит к образованию о– и п-изомеров с преобладанием первого из них:

При нитровании фенола тетраоксидом диазота в инертном растворителе (бензол, дихлорэтан) образуется 2,4-динитрофенол:

Нитрование последнего нитрующей смесью протекает легко и может служить методом синтеза пикриновой кислоты:

Эта реакция идет с саморазогреванием.
Пикриновую кислоту получают также через стадию сульфирования. Для этого обрабатывают фенол при 100°С избыточным количеством серной кислоты и получают 2,4-дисульфопроизводное, которое не выделяя из реакционной смеси обрабатывают дымящей азотной кислотой:

Введение двух сульфогрупп (также как и нитрогрупп) в бензольное ядро делает его устойчивым к окисляющему действию дымящей азотной кислоты; реакция не сопровождается осмолением. Такой метод получения пикриновой кислоты удобен для производства в промышленном масштабе.
Сульфирование. Сульфирование фенола в зависимости от температуры протекает в орто– или пара-положении:

Алкилирование и ацилирование по Фриделю-Крафтсу. Фенолы образуют с хлористым алюминием неактивные соли ArOAlCl2, поэтому для алкилирования фенолов в качестве катализаторов применяют протонные кислоты (H2SO4) или металлооксидные катализаторы кислотного типа (Al2O3). Это позволяет использовать в качестве алкилирующих агентов только спирты и алкены:

Алкилирование протекает последовательно с образованием моно, ди– и триалкилфенолов. Одновременно происходит кислотно-катализируемая перегруппировка с миграцией алкильных групп:

Конденсация с альдегидами и кетонами. При действии щелочных или кислотных катализаторов на смесь фенола и альдегида жирного ряда происходит конденсация в о– и п-положениях. Эта реакция имеет очень большое практическое значение, так как лежит в основе получения важных пластических масс и лаковых основ. При обычной температуре рост молекулы за счет конденсации идет в линейном направлении:



Если реакцию проводить при нагревании, начинается конденсация с образованием разветвленных молекул:

В результате присоединения по всем доступным о– и п-положениям образуется трехмерный термореактивный полимер – бакелит. Бакелит отличается высоким электрическим сопротивлением и термостойкостью. Это один из первых промышленных полимеров.
Реакция фенола с ацетоном в присутствии минеральной кислоты приводит к получению бисфенола:

Последний используют для получения эпоксисоединений.
Реакция Кольбе – Шмидта. Синтез фенилкарбоновых кислот.
Феноляты натрия и калия реагируют с углекислым газом, образуя в зависимости от температуры орто– или пара-изомеры фенилкарбоновых кислот:

Окисление
Фенол легко окисляется под действием хромовой кислоты до п-бензохинона:

Фенолы, содержащие разветвленные алкильные группы в орто– и пара-положении к гидроксильной группе, являются антиоксидантами, стабилизаторами полимерных материалов.
Восстановление
Восстановление фенола в циклогексанон используют для получения полиамида (найлон-6,6).

Отдельные представители
Фенол – кристаллическое вещество с т. пл. 43°С, обладает характерным едким запахом, вызывает ожоги на коже. Это один из первых примененных в медицине антисептиков. Применяется в больших количествах для получения пластических масс (конденсация с формальдегидом), лекарственных препаратов (салициловая кислота и ее производные), красителей, взрывчатых веществ (пикриновая кислота).
Метиловый эфир фенола – анизол – применяется для получения душистых веществ и красителей.
Этиловый эфир фенола – фенетол.
Крезолы (метилфенолы) применяются в производстве пластических масс, красителей, дезинфицирующих средств.
АЛЬДЕГИДЫ И КЕТОНЫ
Лекция 17. НАСЫЩЕННЫЕ АЛЬДЕГИДЫ И КЕТОНЫ
Общая формула. Изомерия. Номенклатура. Способы получения (из спиртов, карбоновых кислот, оксосинтез, из реактивов Гриньяра). Строение карбонильной группы. Сходство С=О и С=С связей. Химические свойства. Реакции присоединения нуклеофильных реагентов. Понятие об общем и специфическом кислотном катализе. Реакции присоединения-отщепления. Альдольная и кротоновая конденсации. Енолизация. Восстановление до спиртов и углеводородов, окисление альдегидов и кетонов.
Альдегиды и кетоны относятся к карбонильным соединениям (содержат группу >С=О) и имеют общую формулу:

Для альдегидов R1=Н, R=Alk, для кетонов R=R1=Alk.
Изомерия альдегидов связана со строением радикалов. Альдегиды или называют по названию тех кислот, в которые они превращаются при окислении (тривиальная номенклатура), или, выбрав за основу самую длинную цепь углеродных атомов, включающую альдегидную группу, прибавляют к ее названию окончание -аль (номенклатура IUPAC). Также альдегиды иногда называют как производные уксусного альдегида:

В префиксе альдегидную группу называют формил-:

Изомерия кетонов связана со строением радикалов и с положением карбонильной группы в углеродной цепи. Кетоны называют по номенклатуре IUPAC аналогично альдегидам, но прибавляют окончание -он. Помимо номенклатуры IUPAC кетоны называют как производные гипотетического соединения – «кетона». Кроме того, широко используются исторически сформировавшиеся (тривиальные) названия:

Способы получения альдегидов и кетонов
Альдегиды и кетоны могут быть получены окислением алкенов (кислородом в присутствии солей палладия и озоном), спиртов и гидратацией алкинов.
Промышленное значение имеет метод гидроформилирования алкенов (оксосинтез).
1. Из спиртов. Дегидрированием спиртов получают многие альдегиды и кетоны, но в настоящее время процесс сохранил свое значение только для получения формальдегида (катализатор Cu):

Промышленным способом получения является окисление спиртов. В качестве окислителей применяют K2Cr2O7/разб. H2SO4, Cr2O3/разб. H2SO4. Окислением первичных спиртов получают альдегиды, вторичных – кетоны.


2. Гидроформилирование алкенов (оксосинтез). Реакция открыта Реленом в 1938 г. (100-200оС, 100-250 атм СО, катализатор – ThO2+mgo на кизельгуре). Процесс (оксосинтез) катализируется дикобальтоктакарбонилом [Co(CО)4]2.

Практическое значение имеют пропионовый, н– и изомасляный альдегиды, получаемые гидроформилированием этилена и пропилена.
3. Восстановление карбоновых кислот и их производных. Прямое восстановление карбоновых кислот для получения альдегидов не применимо, так как альдегиды восстанавливаются легче, чем соответствующие кислоты. Альдегиды можно получать восстановлением хлорангидридов карбоновых кислот (реакция Розенмунда):
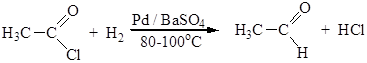
Активность катализатора снижается добавкой контактных ядов, например тетраметилтиомочевины (ТМТМ).
Одним из лучших методов восстановления хлорангидридов кислот является восстановление три-трет-бутоксиалюмогидридом лития при -78°С. Специфический восстановитель и низкая температура исключают дальнейшее восстановление альдегида до спирта.

Восстановитель получают при взаимодействии 1 моля галогенидов лития с 3 молями трет-бутилового спирта.
4. Термическое разложение Са– и Ва-солей карбоновых кислот. Кетоны и альдегиды могут быть получены термическим разложением кальциевых и бариевых солей одноосновных кислот.
Если для реакции берут смесь солей двух кислот или смешанную соль, то протекает реакция между молекулами разных солей. Если же одну из солей – формиат, то получают альдегид:


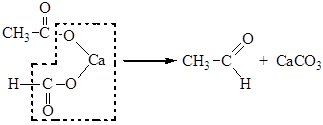
В реакцию могут быть введены непосредственно кислоты. Реакцию проводят при температуре 400-450ºС в присутствии смеси окислов металлов ThO2, MnO, CaO, ZnO.
5. Синтез альдегидов и кетонов с помощью реактивов Гриньяра. При взаимодействии эфиров карбоновых кислот с реактивами Гриньяра происходит присоединение одной молекулы металлорганического соединения к карбонильной группе, гидролиз аддукта приводит к альдегиду или кетону:
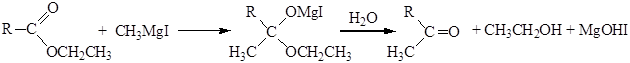
Кетоны получают взаимодействием реактивов Гриньяра с нитрилами:

Альдегиды и кетоны могут быть получены также окислением алкенов (озонолизом), гидратацией ацетилена (уксусный альдегид) и его гомологов (кетоны).
Физические свойства альдегидов и кетонов
Формальдегид – газ с резким запахом, растворим в воде. Водный раствор формальдегида называют формалином. Другие низшие альдегиды и кетоны – жидкости, легко растворимые в воде. Растворимость резко уменьшается с увеличением углеводородного радикала до пяти атомов углерода в молекуле и более. Низшие альдегиды имеют резкий неприятный запах, который при сильном разведении становится приятным (напоминает запах плодов). Высшие гомологи имеют запах цветов и фруктов. Кетоны также обладают сильным запахом.
Плотность альдегидов и кетонов меньше единицы.
При одном и том же составе и строении углеродной цепи кетоны кипят при несколько более высоких температурах, чем альдегиды. Температуры кипения альдегидов и кетонов с нормальным строением углеродной цепи выше, чем соединений изостроения. Альдегиды и кетоны кипят при гораздо более низкой температуре, чем спирты того же состава. Это свидетельствует о слабой диссоциации между молекулами жидкости и отсутствии межмолекулярных водородных связей. В то же время температуры кипения карбонильных соединений значительно выше температур кипения углеводородов с той же молекулярной массой, что связано с их высокой полярностью (табл. 17.1).
Таблица 17.1
Физические свойства альдегидов и кетонов
| Альдегиды | Кетоны | ||||
| Формула | Т. пл. | Т. кип. | Формула | Т. пл. | Т. кип. |
| НСНО | -92 | -21 | - | - | - |
| СН3СНО | -123.5 | - | - | - | |
| С2Н5СНО | -81 | СН3СОСН3 | -95 | ||
| С3Н7СНО | -99 | СН3СОС2Н5 | -86 | ||
| С4Н9СНО | -92 | СН3СОС3Н7 | -78 |
Электронное строение и общая характеристика
реакционной способности
Электронное строение альдегидов и кетонов показано на примере муравьиного альдегида:

Связь углерод-кислород является одновременно более прочной и более реакционноспособной, чем двойная углерод-углеродная связь.
Повышенная реакционная способность связи С=О вызвана различием электроотрицательности углерода и кислорода. В связи с появлением частичного положительного заряда на атоме углерода альдегиды и кетоны проявляют склонность к реакциям с нуклеофильными реагентами. Среди них реакции нуклеофильного присоединения и присоединения-отщепления. Особенности электронного строения служат причиной и других реакций альдегидов и кетонов: протонирование карбонильной группы, СН-кислотность при наличии водородных атомов у α-углеродного атома. Явление СН-кислотности состоит в повышенной подвижности атома водорода в α-положении по отношению к карбонильной группе, вызванной сильным I-эффектом последней:

Электронная плотность от атомов водорода смещена на α-углеродный атом, этим вызвана легкость его замещения. Подвижностью атома водорода в α-положении объясняется и явление кето-енольной таутомерии альдегидов и кетонов:

Кето-енольная таутомерия обуславливает некоторые реакции альдегидов и кетонов, которые будут рассмотрены ниже.
Химические свойства альдегидов и кетонов